First Monkeypox virus sequence from northern Mexico
Kame A. Galán-Huerta 1, Manuel Paz-Infanzon 2, Laura Nuzzolo-Shihadeh 2, Alí F. Ruiz-Higareda 1, Paola Bocanegra-Ibarias 2, Daniel Zacarias-Villarea l3, Luis A. Yamallel-Ortega 3, Maria D. Guerrero-Putz 3, Jorge Ocampo-Candiani 3, Ana M. Rivas-Estilla 1, Adrian Camacho-Ortiz 2
1 Medical Virology Research and Innovation Center, Department of Biochemistry and Molecular
Medicine, School of Medicine, Universidad Autónoma de Nuevo León, Monterrey 64460, Mexico2 Infectious Diseases Service, School of Medicine, University Hospital “Dr. José Eleuterio González”,
Universidad Autónoma de Nuevo León, Monterrey 64460, Mexico3 Dermatology Service, School of Medicine, University Hospital “Dr. José Eleuterio González”,
Universidad Autónoma de Nuevo León, Monterrey 64460, Mexico
Monkeypox virus (MPXV) is a zoonotic pathogen that causes a febrile rash disease in humans. It has caused multiple outbreaks in the past (1,2) but recently gained popularity because of a multi-country outbreak where detected cases had no a clear link to Africa (3).
Monkeypox virus is a double-stranded DNA virus which belongs to the genus Orthopoxvirus, within the Poxviridae family (4). Two genetic clades have been characterized: West African and Central African. However, a new classification was proposed: clades 1, 2 and 3 (5). Genomes belonging to the recent outbreaks gather at clade 3 and create the hMPXV1 subclade. The current international 2022 outbreak is named B.1.
As of the 4th of July 2022, Mexico had 27 confirmed monkeypox infections (6). We report a complete genome of the first monkeypox case detected in Nuevo León, Mexico.
A skin swab of the lesions was collected on June 28th, 2022, at Dermatology Service of the University Hospital “Dr. José Eleuterio González”, from a 34 years-old male patient. Viral DNA was extracted from with the High Pure PCR Template Preparation Kit (Roche) in a biological safety cabinet and all the hazardous biological infectious wastes were disposed according to the NOM-087-ECOL-SSA1-2002.
Monkeypox virus was detected by qPCR using the protocol reported by Li (7). DNA was quantified and 700 ng were used to prepare the library with SQK-LSK109, following the manufacturer’s protocol. The library was loaded on a FLO-MIN106 R9.4.1. We used high accuracy basecalling and deplete the human genome by adaptive sampling reference (GRCh38.p14, GCF_000001405.40) in MinKNOW v.22.05.5. The run lasted for 20 h and obtained 84,000 reads.
The genome was assembled with Minimap2 v. 2.17 using a Monkeypox virus reference genome (NC_063383.1) and consensus sequence was obtained with Medaka v. 1.0.3 and bcftools v.1.10.12. The obtained sequence was deposited in GISAID (EPI_ISL_13607904) and GenBank (ON911481). We obtained the complete sequence of Monkeypox virus (197,667 bp). The sequence comprised 4574 mapped reads, 78x average depth and 128x maximum coverage depth.
We downloaded 144 sequences from Genbank that belonged to clade 3 and hMPXV1 from 2017 - 2019 and 2022 outbreaks. Sequences were aligned in MAFFT v.7.505 8, and the product was used to infer a maximum likelihood phylogenetic tree. The tree was inferred with IQ-TREE v.2.0.3 9 using the HKY+F+I substitution model with 1000 replicates for ultrafast bootstrap and 1000 replicates for SH approximate likelihood ratio test. The tree was visualized in FigTree v.1.4.4 and annotated with ggtree v.3.4.0 10 and treeio v.1.20. 11
The newly obtained sequence groups within the newly proposed B.1 lineage from Clade 3, where hMPXV1 are located. Sequences from the multi-country monkeypox outbreak are in lineage B.1. The obtained sequence groups in a moderate-supported (SH_aLRT 77, UFboot 90) clade with sequences from Germany.
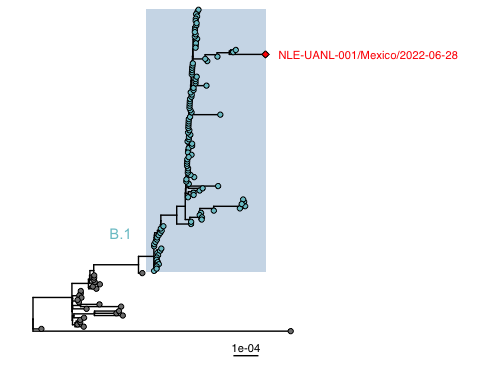
Figure. Maximum likelihood phylogenetic tree with 145 whole genome sequences. Highlighted genomes in blue belong to the proposed B.1 lineage. The genome described in this study is indicated with a red diamond. The list of accession numbers of the sequences used in this alignment is shown in supplementary table 1.
Acknowledgments
This work was funded by Consejo Nacional de Ciencia y Tecnología (CONACyT) with the following grant number 312328, awarded to Kame A. Galán-Huerta and 315848, awarded to Ana M. Rivas-Estilla.
References
-
Ladnyj ID, Ziegler P, Kima E. A human infection caused by monkeypox virus in Basankusu Territory, Democratic Republic of the Congo. Bull World Health Organ. 1972;46(5):593.
-
Yinka-Ogunleye A, Aruna O, Dalhat M, et al. Outbreak of human monkeypox in Nigeria in 2017-18: a clinical and epidemiological report. Lancet Infect Dis. 2019;19(8):872-879.
-
Vivancos R, Anderson C, Blomquist P, et al. Community transmission of monkeypox in the United Kingdom, April to May 2022. Euro Surveill. 2022;27(22).
-
World Health Organization. Monkeypox. Published 2022. Accessed June 30, 2022. Mpox (monkeypox)
-
Happi C, Adetifa I, Mbala P, et al. Urgent need for a non-discriminatory and non-stigmatizing nomenclature for monkeypox virus. Virological.org. Published 2022. Accessed July 4, 2022. https://pando.tools/t/urgent-need-for-a-non-discriminatory-and-non-stigmatizing-nomenclature-for-monkeypox-virus/853
-
Panamerican Health Organization, World Health Organization. Monkeypox cases - Region of the Americas. Published 2022. Accessed July 5, 2022. https://shiny.pahobra.org/monkeypox/
-
Li Y, Zhao H, Wilkins K, Hughes C, Damon IK. Real-time PCR assays for the specific detection of monkeypox virus West African and Congo Basin strain DNA. J Virol Methods. 2010;169(1):223-227.
-
Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. Published online September 6, 2017.
-
Minh BQ, Schmidt HA, Chernomor O, et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol. 2020;37(5):1530-1534.
-
Yu G, Smith DK, Zhu H, Guan Y, Lam TTY. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8(1):28-36.
-
Wang LG, Lam TTY, Xu S, et al. Treeio: An R Package for Phylogenetic Tree Input and Output with Richly Annotated and Associated Data. Mol Biol Evol. 2020;37(2):599-603.