Emergence of SARS-CoV-2 lineage A.27 in Germany, expressing viral spike proteins with several amino acid replacements of interest, including L18F, L452R, and N501Y in the absence of D614G
Sébastien Calvignac-Spencer1*, Matthias Budt2, Matthew Huska3, Hugues Richard3, Max von Kleist4, Stefan Kröger5, Torsten Semmler6, Thorsten Wolff2, Martin Hölzer3*
1 Epidemiology of Highly Pathogenic Microorganisms, Robert Koch Institute, Berlin
2 Influenza and other Respiratory Viruses, Robert Koch Institute, Berlin
3 Methodology and Research Infrastructure, Bioinformatics, Robert Koch Institute, Berlin
4 Systems Medicine of Infectious Disease, Robert Koch Institute, Berlin
5 Infectious Disease Epidemiology, Robert Koch Institute, Berlin,
6 Methodology and Research Infrastructure, Genome Sequencing and Genomic Epidemiology, Robert Koch Institute, Berlin
*corresponding authors [email protected], [email protected]
Summary
Here, we report on the increasing frequency of the SARS-CoV-2 lineage A.27 in Germany during the first months of 2021. Genomic surveillance at the Public Health Institute in Germany identified 710 A.27 genomes as of May 02nd, 2021, with a vast majority identified in laboratories from a single German state (Baden-Wuerttemberg, n=572; 80.5%). Baden-Wuerttemberg is located near the border with France, from where to date most A.27 sequences were submitted to GISAID. The first appearance of this lineage based on sequencing in a laboratory in Baden-Wuerttemberg can be dated to early January. From then on, the relative abundance of A.27 increased until the end of February but has since declined - meanwhile, the abundance of B.1.1.7 increased in the region. The A.27 lineage shows a mutational pattern typical of VOIs/VOCs, including an accumulation of amino acid substitutions in spike. Among those, L18F, L452R and N501Y are located in the N-terminal- (NTD) or receptor binding domain (RBD) epitope regions and have been suggested to result in immune escape and higher transmissibility. In addition, A.27 does not show the D614G mutation typical for all VOIs/VOCs from the B lineage. Overall, it seems sensible to further monitor the spread of A.27 nationally and internationally, although the observed trend in Baden-Wuerttemberg suggests that it will also be outcompeted by B.1.1.7.
Results & Discussion
Increase of A.27 cases in Baden-Wuerttemberg, Germany
A total of 377 A.27 genomes from 16 different (mostly European) countries have been made available on GISAID (as of May 10th, 2021). A.27 was first detected in Denmark in mid-December, however, 214 genomes were reported from France including 12 from Mayotte, an overseas region and single territorial collectivity of France. The numbers might therefore point to an origin of A.27 in France where the lineage may have spread early.
Genomic surveillance at the Robert Koch Institute (RKI, Public Health Institute Germany) identified 710 sequences belonging to the lineage A.27 (as of May 02nd, 2021). The earliest sequence was discovered in a laboratory in Baden-Wuerttemberg (BW) and occurred in calendar week (CW) 01 in 2021; since then, a vast majority of A.27 cases in Germany were sequenced in BW (n=572 to CW16, based on data through May 02nd, 2021). Based on sequencing, the relative abundance of this lineage in the region increased until CW08 (6.12%) but has since then decreased again (1.21% in CW13), see Tab. 1. Meanwhile, the frequency of the VOC B.1.1.7 kept increasing in the region (5.84% in CW 03, 71 % in CW 14; data not shown). The number of detected A.27 whole-genome sequences until May 02nd, 2021 that were either submitted to the RKI via the German Electronic Sequencing Data Hub (DESH, n=707) or directly sequenced at the RKI as part of the SARS-CoV-2 Integrated Molecular Surveillance lab-network (IMS-SC2, n=3) is shown in Tab. 1.
| CW01 | 02 | 03 | 04 | 05 | 06 | 07 | 08 | 09 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 2021 | |
| GER | 3 (1.07) | 2 (0.22) | 7 (0.32) | 29 (0.82) | 40 (0.92) | 53 (0.89) | 61 (0.91) | 96 (1.35) | 79 (1.02) | 84 (0.91) | 86 (0.81) | 65 (0.62) | 52 (0.48) | 27 (0.25) | 18 (0.14) | 8 (0.10) | 710 |
| BW | 3 (2.75) | 0 (0.00) | 6 (1.34) | 28 (4.21) | 35 (4.69) | 52 (4.47) | 52 (4.12) | 83 (6.12) | 76 (3.98) | 54 (2.06) | 53 (1.72) | 51 (1.64) | 47 (1.21) | 21 (0.59) | 9 (0.29) | 2 (0.22) | 572 |
| SH | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 2 (0.60) | 3 (1.07) | 0 (0.00) | 22 (6.02) | 23 (5.37) | 4 (0.99) | 0 (0.00) | 2 (0.51) | 3 (0.67) | 1 (0.43) | 60 |
Table continued:
| CW12 | 13 | 14 | 15 | 16 | 2021 | |
| GER | 65 (0.62) |
52 (0.48) |
27 (0.25) |
18 (0.14) |
8 (0.10) |
710 |
| BW | 51 (1.64) |
47 (1.21) |
21 (0.59) |
9 (0.29) |
2 (0.22) |
572 |
| SH | 4 (0.99) |
0 (0.00) |
2 (0.51) |
3 (0.67) |
1 (0.43) |
60 |
Table 1. The number of detected A.27 whole-genome sequences at the RKI per calendar week (CW) based on data collected until May 02nd, 2021 and shown up to CW16 (random and suspect sampling combined). Relative frequencies (in %) in relation to all sequences from the respective CW are given in parentheses. Later time periods may be biased due to missing data. GER – Germany, BW – Baden-Wuerttemberg, SW – Schleswig-Holstein.
Of the 710 A.27 genomes from Germany, 206 were obtained following a random sampling strategy while 271 were collected based on suspect samples (remainder unknown). For sequences from both categories (suspect, random), the proportion of reported A.27 sequences was compared between calendar weeks and we searched for a significant increase (Fisher exact test, adjusted p-value < 0.1, see Methods). In CW 07-08, we observed a 2.1-fold increase in proportion among the suspect samples in the BW region, followed the next week by a 3-fold increase on the set of samples obtained following a random sampling strategy (Fig. 1A). Interestingly, on the random samples and during the same period CW 07-10, Schleswig-Holstein also shows an impressive 33-fold increase in proportion (Fig. 1A) but absolute numbers are still low (Tab. 1). Please also note that no A.27 sequences were detected in CW08 and 09 for that region.
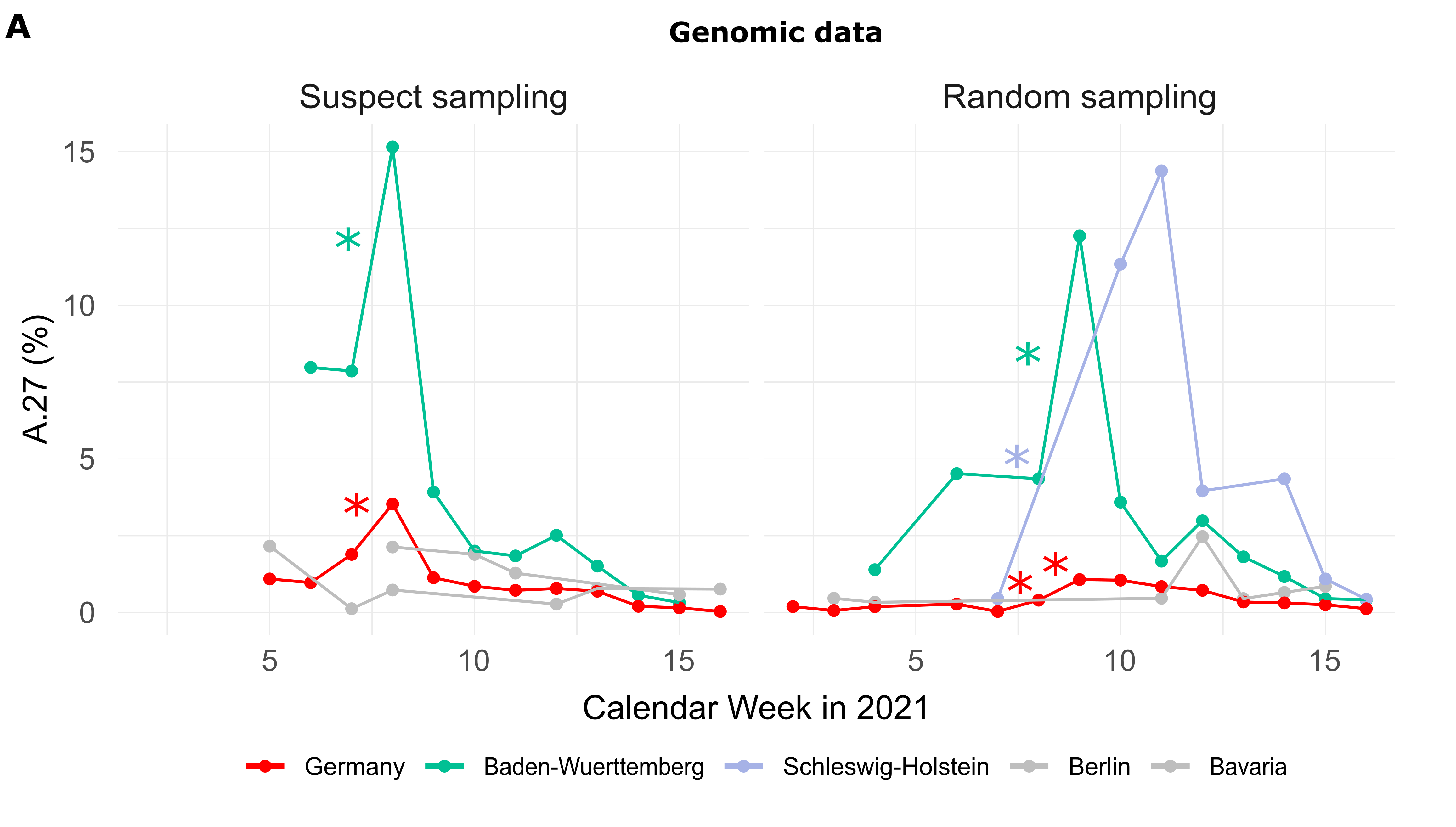

Figure 1. (A) Proportion of reported A.27 sequences compared between calendar weeks (CW) and based on suspect (n=271) and random sampling (n=206) strategies. Significant increases in proportion are marked with a star (adjusted p-value < 0.1). In CW 07-08, we observed a 2.1-fold increase in the proportion among suspect samples in the BW region and in CW 08-09 among random samples in the same region. Between CW07 and CW10 we observed an impressive 33-fold increase in proportion in Schleswig-Holstein among the random samples, however, no A.27 sequences were detected in CW08 and 09 for that region, and absolute numbers are low. (B) Epidemiological data of reported cases of lineage A.27 (n=205) in Germany per age group. Median age of cases was 45 years. Data points with missing information were set to zero. In general, no data were available for CW04-06 as of April 30th, 2021. (C) Distribution of cases over federal states based on epidemiological data, 92% were notified in Baden-Wuerttemberg (date of reporting: April 30th, 2021) Epidemiological data was not available for all federal states. (download high-res PDF)
Epidemiological data
For 205 cases reported between CW03 and 14 additional information was available via the national electronic reporting system for surveillance of notifiable infectious diseases (SurvNet, implemented in 2001; (Krause et al. 2007)) (Fig. 1B and C; geographic distribution similar to that seen in the entire dataset, data not shown). Most cases were reported for middle-aged patients (Fig. 1B). Hospitalization was reported in 17 patients, and three died. None of the 205 reported cases with epidemiologic data originated in Schleswig-Holstein, underscoring the importance of integrating multiple data sources for effective molecular surveillance.
Phylogenetic analysis of A.27 genomes
All A.27 genomes appeared in a monophyletic group whose most recent common ancestor (MRCA) was dated to early August 2020 (2020-08-06; Fig. 2). However, all but three basal A.27 sequences formed a clade whose time to MRCA was much more recent, lying around late October 2020 (2020-10-29). We also observe two A.27 clades in the tree, only one of which is more closely associated with sequences originating in France (Fig. 2).
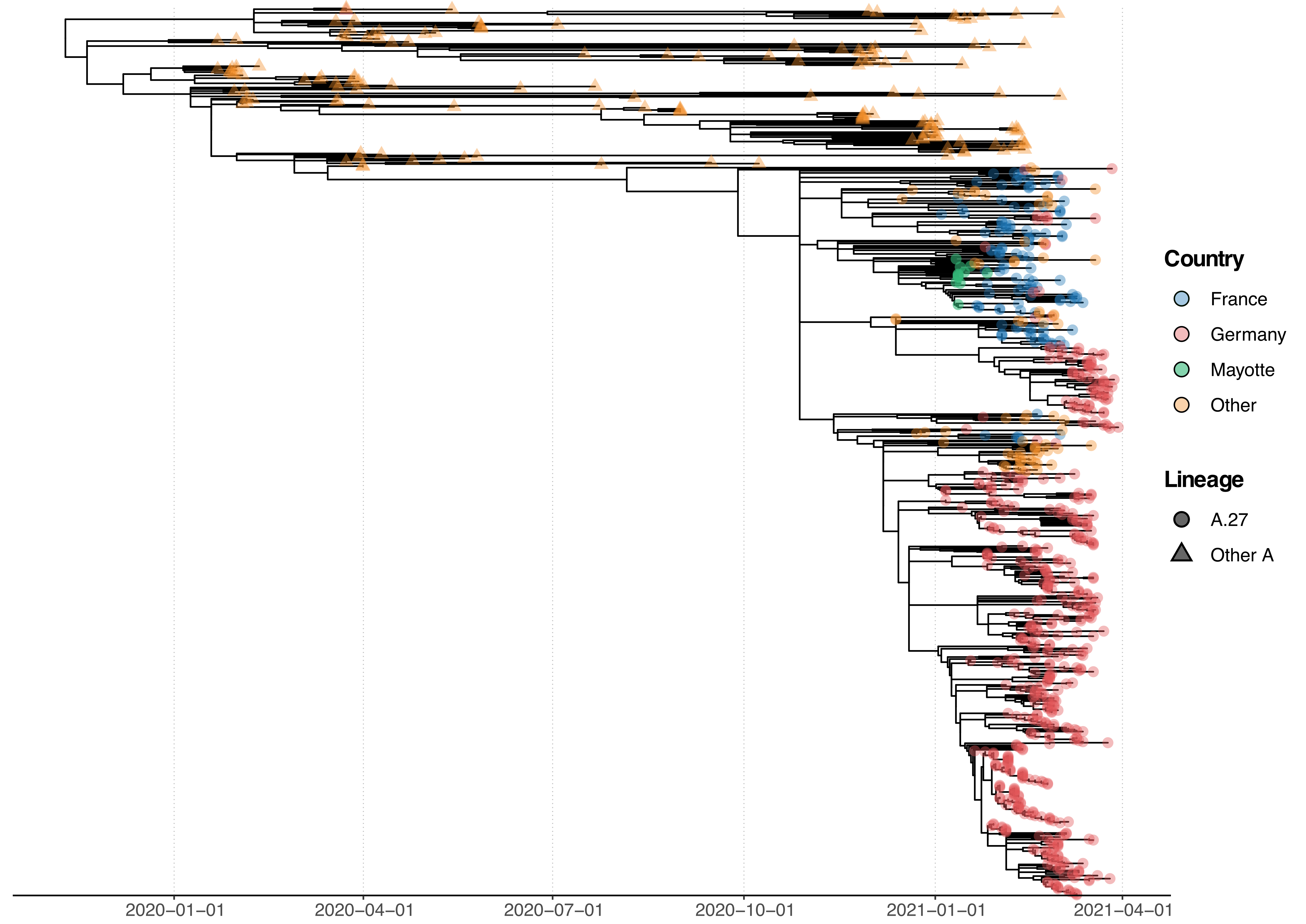
Figure 2. Phylogenetic analysis of A.27 genomes forming a monophyletic group. We also observed three basal A.27 genomes missing some of the seven characteristic A.27 spike mutations and showing no deletions in spike. (download high-res PDF)
Spike mutations of interest and concern in A.27
The A.27 lineage is characterized by a high number of characteristic (lineage-defining) mutations that are also known from other VOC/VOI (Fig. 3C). In total, A.27 harbors 17 lineage-defining mutations (Fig. 3A and Fig. 4A), seven of which result in non-synonymous nucleotide substitutions in the spike protein: L18F, L452R, N501Y, A653V, H655Y, D796Y, G1219V. Among those, three alterations are of particular concern as they are located in the NTD or RBD epitope regions, respectively, and are suspected to result in immune escape (L18F, L452R and N501Y) and/or higher infectivity and transmissibility (L452R and N501Y). Antibody escape data for the RBD, integrating multiple experimental studies from the Bloom lab (Greaney et al. 2021) (https://jbloomlab.github.io/SARS2_RBD_Ab_escape_maps, accessed May 03rd, 2021), assign maximum mutation escape scores of 0.97 (L452R) and 0.90 (N501) over multiple antibodies/sera or antibody/serum types.
The polymorphic positions are labeled on the S protein structure (Fig. 3B), indicating the localization of L452R (reduced antibody neutralization (Deng et al. 2021; Li et al. 2020)) and N501Y (related to increased transmissibility (Liu et al. 2021)) in the RBD, A653V/H655Y in proximity to the S1/S2 furin cleavage site at position 681 that promotes infection and cell-cell fusion (Papa et al. 2021), and D795Y, closely located to the essential TMPRSS2-cleavage site (Bestle et al. 2020) at position 815.
The L18F replacement is part of an antigenic supersite in the N-terminal domain of the spike protein (McCallum et al. 2021). Antibodies targeting NTD were shown to be less abundant, but more potent than those targeting RBD (McCallum et al. 2021). Mutations at this position were selected via in vitro passaging experiments and showed cross-resistance to monoclonal antibodies targeting NTD (McCallum et al. 2021). The S-L18F alteration also appears in VOCs that are associated with immune escape and reinfection, such as B.1.351 and P.1. Recently, the L452R change in the RBD also known from B.1.427/429 (Shen et al. 2021), B.1.526.1 and B.1.617 sub-lineages (Fig. 3C) was suggested to contribute to escaping human leukocyte antigen-restricted cellular immunity while also increasing the binding affinity to ACE2, thus, increasing viral infectivity and potentially enhancing virus replication (Motozono et al. 2021). S-N501Y also appears in VOC’s B.1.1.7, P.1 and B.1.351, and may increase infectivity in vitro (Kuzmina et al. 2021) and appears to confer resistance against some RBD targeting antibodies (class 1) (Wang et al. 2021). The H655Y substitution adjacent to the S1/S2 cleavage site and also known from P.1 was recently detected in a potential new VOI within the A lineage (temporarily designated A.VOI.V2) identified from three cases of incoming travelers from Tanzania to Angola (de Oliveira et al. 2021).
Importantly, A.27 generally does not possess mutation D614G, which emerged early in the pandemic and rapidly became dominant. Mutation D614G is associated with increased receptor binding (Ozono et al. 2021) and infectivity (Zhang et al. 2020). Neither does A.27 contain the likely functionally equivalent Q613H that is present in A.23.1 (Bugembe et al. 2021) (Fig. 3C). The absence of D614G may indicate an early separation of the lineage from the globally circulating variants with an endemic evolution. Moreover, it could point towards overall lower fitness of A.27 in comparison to major circulating variants like B.1.1.7. In line with this presumption, the overall frequency of A.27 is declining. The presence of apparent escape mutants L18F and L452R might indicate nascent immune evasion.
Almost all A.27 genomes have multiple deletions such as ORF3a:del258/259 and ORF8:del119/120, a few of which are highly conserved (Fig. 3 and 4). We also observed in 58 (8.07 %) of the investigated A.27 genomes a 12 nt long insertion (CTTTCGATCTCT) located at position 27,373 near the 3’-end of ORF6. However, all sequences with this insertion were submitted by a single laboratory via the German Electronic Sequencing Data Hub (DESH) and further investigation showed that the inserted sequence is similar to a potential primer sequence in the first genomic amplicon. Therefore, we assume a technical error in the sequencing protocol or in the genome reconstruction pipeline of this laboratory.
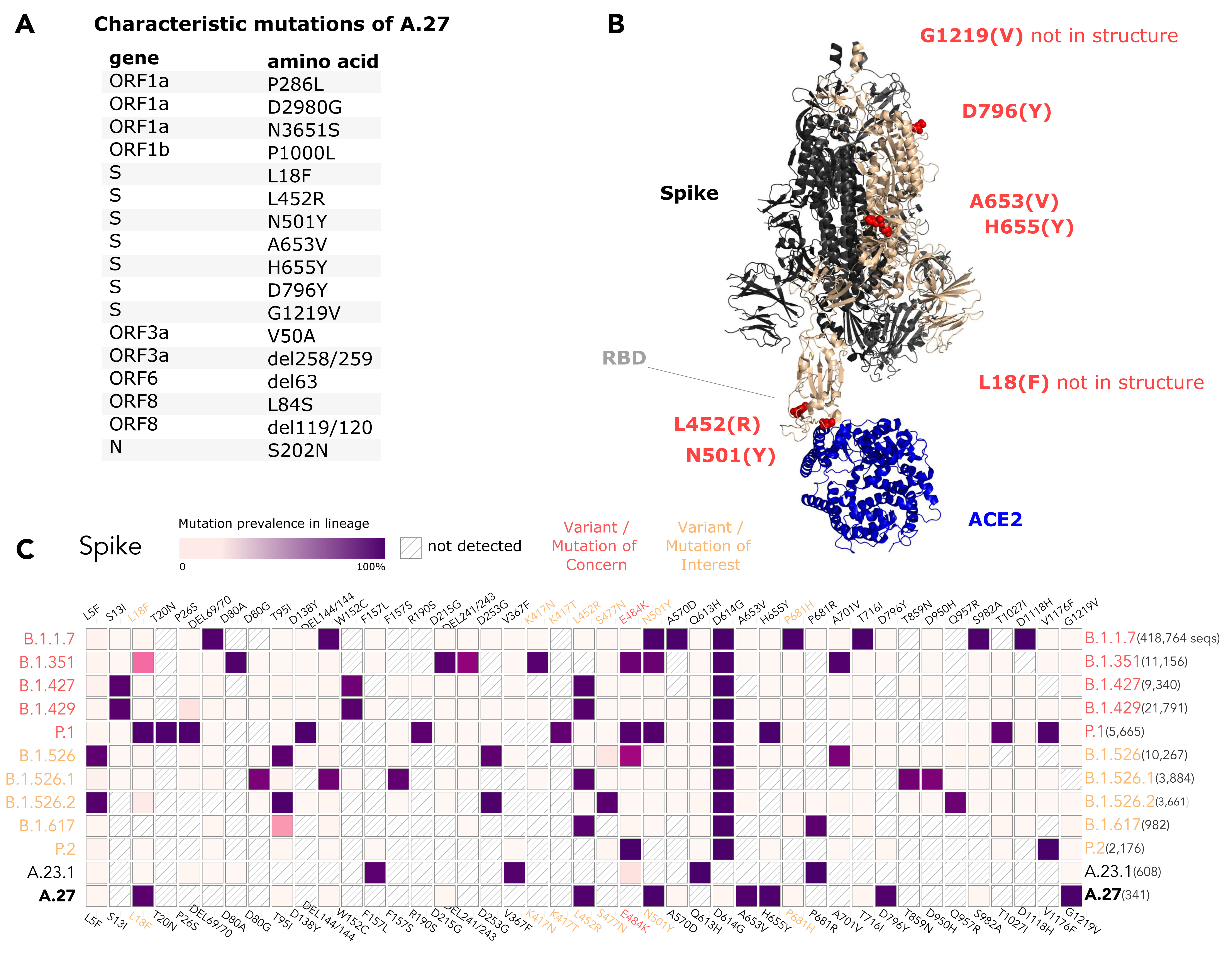
Figure 3. (A) Characteristic (lineage-defining) mutations of A.27 derived from https://outbreak.info. (B) The trimeric spike protein of SARS-CoV-2 (PDB: 7kj2, (DeLano 2002)) is depicted in complex with its cellular receptor ACE2. The variant residues of lineage A.27 are labeled on one subunit with the A.27 amino acids indicated in parentheses. RBD - receptor binding domain. (C) Mutation profiles of selected VOC/VOI in comparison to A.27 and A.23.1 obtained from https://outbreak.info/ (April 25th 2021), (Mullen et al. 2021b). (download high-res PDF)
Spike mutations of interest and concern in basal A.27 lineages
The rare, relatively deep-branching lineages in the phylogenetic tree (Fig. 2) prompted us to check the mutation profile of the corresponding three genomes (Fig. 4, GISAID accessions: EPI_ISL_1170076, EPI_ISL_1567985, EPI_ISL_1353586). None possessed all the seven aforementioned non-synonymous mutations in spike, nor any of the frequently observed deletions (Fig. 4). Intriguingly, one of the genomes (EPI_ISL_1567985) encoded S-L452R, S-N501Y, and S-G1219V associated with six other amino acid changes in the spike (A570D, D614G, P681H, T716I, S982A, I1221V), most interestingly comprising D614G and also P681H (Fig. 4B). While we report here on these three unusual A.27 genomes, further analysis is required also on their raw sequencing data to confirm the observed changes that distinguish these sequences from standard A.27. Please also note that we were not able to upload EPI_ISL_1353586 to GISAID due to an incorrectly masked deletion at position 28,252 causing a frameshift.
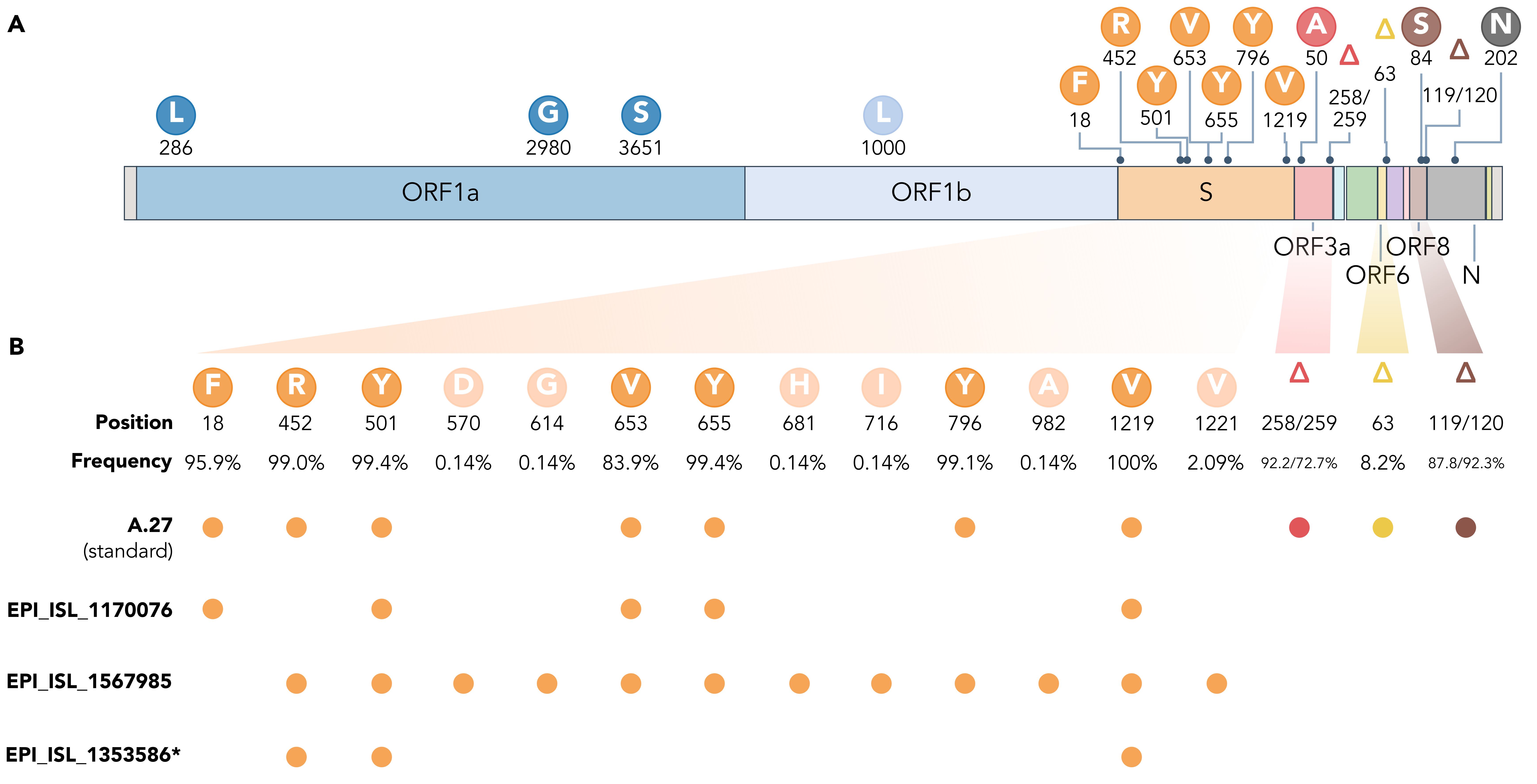
Figure 4. (A) Characteristic mutations leading to 17 amino acid alterations or deletions in A.27 genomes with a prevalence of at least 75%. Figure obtained and adjusted from (Mullen et al. 2021a). (B) Overview of spike substitutions and high-frequency deletions in ORF3a, ORF6, and ORF8 in genomes assigned the A.27 lineage via pangolin. Frequencies are calculated based on the set of 718 A.27 genomes as used for the tree in Fig. 2. Among “standard” A.27 genomes harboring all seven spike mutations and the three prevalent deletions, we also found three genomes that show a varying mutation profile in the spike and missing all three high-frequency deletions. Among them, EPI_ISL_1567985 shows a high frequency of spike mutations and, in particular, D614G. While the deletion on ORF6 at position 63 is listed with a high frequency (higher 75%) on outbreak.info, we only observe a frequency of 8.2% in our data set based on detection via NextStrain. *EPI_ISL_1353586 is not yet available on GISAID due to rejection of the sequence because of a frameshift. (download high-res PDF)
Conclusion
Here, we report the increasing frequency of the SARS-CoV-2 lineage A.27 in Germany, particularly in Baden-Wuerttemberg (BW), from January to February 2021. Since this lineage shows mutations associated with both higher transmissibility and reduced antibody reactivity, we suggest that enhanced monitoring is warranted on both national and international levels. The A.27 lineage was indeed continuously detected in other European countries (France, Slovenia or the United Kingdom). However, the ongoing trend in BW indicates that A.27 may be outcompeted by B.1.1.7. Based on genomic data obtained via a random sampling strategy, we also found a significant increase in the proportion of A.27 sequences between CW 07-10 in Schleswig-Holstein, highlighting the importance of integrating genomic and epidemiological data sources for effective molecular surveillance. Finally, our analyses identify rare but clearly divergent genomes assigned to A.27, suggesting the current definition of the lineage probably needs to be reexamined. One of these rare genomes combines a D614G background and several mutations of concern and should therefore also be monitored.
Methods
Detecting increase in proportion
Based on the genomic data, we tested for differences in the proportion of the A.27 lineage using a Fisher exact test. Tests were performed separately for suspect and random sampling strategies. For each German state, the test was performed on a 2x2 count table showing, for pairs of consecutive calendar weeks (CW), the number of A.27 samples and the total number of non-A.27 samples in a state. If no A.27 sequences were identified for a particular federal state in a given week, that week was skipped and sequences from the next week for that state were considered instead. We chose this approach to be more conservative in detecting an increase in the proportion. Only states in which A.27 samples were detected in at least three CW were considered. The obtained set of p-values was then corrected for multiple testing using a Benjamin-Hochberg procedure, keeping the adjusted p-values below 0.1.
Phylogenetic reconstruction
We used all available A.27 genomes and the selection of A genomes from NextStrain (Hadfield et al. 2018) global build to assemble a dataset for phylogenetic analyses including 718 A.27 genomes from RKI and GISAID. After excluding low quality/missing sampling date genomes (<90% reference genome coverage, >10 non-ACGTN ambiguous positions, >40 SNPs with respect to NC_045512.2), we aligned a total of 875 genomes with MAFFT using default parameters (Katoh and Standley 2013). We used the resulting alignment to estimate a no-clock maximum-likelihood tree with IQ-TREE2 (Minh et al. 2020). This tree showed a strong temporal signal in a regression of root-to-tip distance versus time (Rsq: 0.81) and was therefore used to estimate a time tree using tip dates only with TreeTime (Sagulenko, Puller, and Neher 2018).
Three-dimensional structure of the S protein
The PyMOL software version 1.7.2.1 (DeLano 2002) was used to label variant residues on the structure of the spike protein based on the template structure 7kj2 obtained from the Protein Data Base (www.pdb.org) and provided by (Xiao et al. 2021).
Acknowledgment
We gratefully acknowledge the authors from the originating laboratories responsible for obtaining the specimens and the submitting laboratories where genetic sequence data were generated and shared via DESH (German Electronic Sequencing Data Hub) and GISAID, on which this research is based. We thank MF4 and the DESH team for data handling and sharing and MF2 and P3 for in-house sequencing in the context of the Integrated Molecular Surveillance SARS-CoV-2 (IMS-SC2). We thank all MF1 team members involved in SARS-CoV-2 data handling, analyses, and reporting and particularly Oliver Drechsel for dealing with GISAID data uploads and Stephan Fuchs for fruitful discussions. We thank Jonas Fuchs for a helpful exchange, especially about the likely sequence insertion artifact found in a subset of the A.27 genomes.
References
Bestle, Dorothea, Miriam Ruth Heindl, Hannah Limburg, Thuy Van Lam van, Oliver Pilgram, Hong Moulton, David A. Stein, et al. 2020. “TMPRSS2 and Furin Are Both Essential for Proteolytic Activation of SARS-CoV-2 in Human Airway Cells.” Life Science Alliance 3 (9). https://doi.org/10.26508/lsa.202000786.
Bugembe, Daniel Lule, My V.T.Phan, Isaac Ssewanyana, Patrick Semanda, Hellen Nansumba, Beatrice Dhaala, Susan Nabadda, et al. 2021. “A SARS-CoV-2 Lineage A Variant (A.23.1) with Altered Spike Has Emerged and Is Dominating the Current Uganda Epidemic.” BioRxiv. medRxiv. https://doi.org/10.1101/2021.02.08.21251393.
DeLano, W. L. 2002. “Pymol: An Open-Source Molecular Graphics Tool.” CCP4 Newsletter on Protein Crystallography. http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.231.5879&rep=rep1&type=pdf#page=44.
Deng, Xianding, Miguel A. Garcia-Knight, Mir M. Khalid, Venice Servellita, Candace Wang, Mary Kate Morris, Alicia Sotomayor-González, et al. 2021. “Transmission, Infectivity, and Antibody Neutralization of an Emerging SARS-CoV-2 Variant in California Carrying a L452R Spike Protein Mutation.” MedRxiv : The Preprint Server for Health Sciences, March. https://doi.org/10.1101/2021.03.07.21252647.
Greaney, Allison J., Tyler N. Starr, Pavlo Gilchuk, Seth J. Zost, Elad Binshtein, Andrea N. Loes, Sarah K. Hilton, et al. 2021. “Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain That Escape Antibody Recognition.” Cell Host & Microbe 29 (1): 44-57.e9.
Hadfield, James, Colin Megill, Sidney M. Bell, John Huddleston, Barney Potter, Charlton Callender, Pavel Sagulenko, Trevor Bedford, and Richard A. Neher. 2018. “Nextstrain: Real-Time Tracking of Pathogen Evolution.” _Bioinformatics _ 34 (23): 4121–23.
Katoh, Kazutaka, and Daron M. Standley. 2013. “MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability.” Molecular Biology and Evolution 30 (4): 772–80.
Krause, Gérard, Doris Altmann, Daniel Faensen, Klaudia Porten, Justus Benzler, Thomas Pfoch, Andrea Ammon, Michael H. Kramer, and Hermann Claus. 2007. “SurvNet Electronic Surveillance System for Infectious Disease Outbreaks, Germany.” Emerging Infectious Diseases 13 (10): 1548–55.
Kuzmina, Alona, Yara Khalaila, Olga Voloshin, Ayelet Keren-Naus, Liora Boehm-Cohen, Yael Raviv, Yonat Shemer-Avni, Elli Rosenberg, and Ran Taube. 2021. “SARS-CoV-2 Spike Variants Exhibit Differential Infectivity and Neutralization Resistance to Convalescent or Post-Vaccination Sera.” Cell Host & Microbe 29 (4): 522-528.e2.
Li, Qianqian, Jiajing Wu, Jianhui Nie, Li Zhang, Huan Hao, Shuo Liu, Chenyan Zhao, et al. 2020. “The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity.” Cell 182 (5): 1284-1294.e9.
Liu, Yang, Jianying Liu, Kenneth S. Plante, Jessica A. Plante, Xuping Xie, Xianwen Zhang, Zhiqiang Ku, et al. 2021. “The N501Y Spike Substitution Enhances SARS-CoV-2 Transmission.” BioRxiv. https://doi.org/10.1101/2021.03.08.434499.
McCallum, Matthew, Anna De Marco, Florian A. Lempp, M. Alejandra Tortorici, Dora Pinto, Alexandra C. Walls, Martina Beltramello, et al. 2021. “N-Terminal Domain Antigenic Mapping Reveals a Site of Vulnerability for SARS-CoV-2.” Cell, March. Redirecting.
Minh, Bui Quang, Heiko A. Schmidt, Olga Chernomor, Dominik Schrempf, Michael D. Woodhams, Arndt von Haeseler, and Robert Lanfear. 2020. “IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era.” Molecular Biology and Evolution 37 (5): 1530–34.
Motozono, Chihiro, Mako Toyoda, Jiri Zahradnik, Terumasa Ikeda, Akatsuki Saito, Toong Seng Tan, Isaac Ngare, et al. 2021. “An Emerging SARS-CoV-2 Mutant Evading Cellular Immunity and Increasing Viral Infectivity.” BioRxiv. https://doi.org/10.1101/2021.04.02.438288.
Mullen, Julia L., Ginger Tsueng, Alaa Abdel Latif, Manar Alkuzweny, Marco Cano, Emily Haag, Jerry Zhou, et al. 2021a. “A.27 Lineage Report from Outbreak.Info.” 2021. outbreak.info SARS-CoV-2 data explorer.
———. 2021b. “SARS-CoV-2 Lineage Comparison from Outbreak.Info.” 2021. outbreak.info SARS-CoV-2 data explorer.
Oliveira, Tulio de, Silvia Lutucuta, John Nkengasong, Joana Morais, Joana Paula Paixão, Zoraima Neto, Pedro Afonso, et al. 2021. “A Novel Variant of Interest of SARS-CoV-2 with Multiple Spike Mutations Detected through Travel Surveillance in Africa.” BioRxiv. medRxiv. https://doi.org/10.1101/2021.03.30.21254323.
Ozono, Seiya, Yanzhao Zhang, Hirotaka Ode, Kaori Sano, Toong Seng Tan, Kazuo Imai, Kazuyasu Miyoshi, et al. 2021. “SARS-CoV-2 D614G Spike Mutation Increases Entry Efficiency with Enhanced ACE2-Binding Affinity.” Nature Communications 12 (1): 848.
Papa, Guido, Donna L. Mallery, Anna Albecka, Lawrence G. Welch, Jérôme Cattin-Ortolá, Jakub Luptak, David Paul, et al. 2021. “Furin Cleavage of SARS-CoV-2 Spike Promotes but Is Not Essential for Infection and Cell-Cell Fusion.” PLoS Pathogens 17 (1): e1009246.
Sagulenko, Pavel, Vadim Puller, and Richard A. Neher. 2018. “TreeTime: Maximum-Likelihood Phylodynamic Analysis.” Virus Evolution 4 (1): vex042.
Shen, Xiaoying, Haili Tang, Rolando Pajon, Gale Smith, Gregory M. Glenn, Wei Shi, Bette Korber, and David C. Montefiori. 2021. “Neutralization of SARS-CoV-2 Variants B.1.429 and B.1.351.” The New England Journal of Medicine, April. https://doi.org/10.1056/NEJMc2103740.
Wang, Zijun, Fabian Schmidt, Yiska Weisblum, Frauke Muecksch, Christopher O. Barnes, Shlomo Finkin, Dennis Schaefer-Babajew, et al. 2021. “MRNA Vaccine-Elicited Antibodies to SARS-CoV-2 and Circulating Variants.” Nature 592 (7855): 616–22.
Xiao, Tianshu, Jianming Lu, Jun Zhang, Rebecca I. Johnson, Lindsay G. A. McKay, Nadia Storm, Christy L. Lavine, et al. 2021. “A Trimeric Human Angiotensin-Converting Enzyme 2 as an Anti-SARS-CoV-2 Agent.” Nature Structural & Molecular Biology 28 (2): 202–9.
Zhang, Lizhou, Cody B. Jackson, Huihui Mou, Amrita Ojha, Haiyong Peng, Brian D. Quinlan, Erumbi S. Rangarajan, et al. 2020. “SARS-CoV-2 Spike-Protein D614G Mutation Increases Virion Spike Density and Infectivity.” Nature Communications 11 (1): 6013.