Felipe Gomes Naveca1,2 Tatiana Amaral Pires de Almeida1,3, Victor Souza1, Valdinete Nascimento1, Dejanane Silva1, Fernanda Nascimento1, Matilde Mejía1, Yasmin Silva de Oliveira1, Luisa Rocha4, Natana Xavier4, Janis Lopes4, Rodrigo Maito4, Cátia Meneses4, Tatyana Amorim5, Daniel Barros5, Luciana Fé5, Ana Ruth Arcanjo5, Guilherme Araújo5, Rosiane Rodrigues6, Stella Albuquerque6, Cristiane Mattos6, Ciciléia Silva6, Aline Linhares6, Taynã Rodrigues7, Francy Mariscal7, Karolina Sabino7, Selso Lopes7, Leandro Cavalcante7, Josilene Rocha7, Márcia Andréa Morais8, Anne Paiva9, Karina Ribeiro9, Deusilene Vieira10, Regina Maria Pinto de Figueiredo11, Ágata Rossi12, Ighor Arantes2, Tiago Gräf13, Túlio Campos14, Gabriel Wallau14, Ana Cecília Ribeiro Cruz15, Livia Neves Casseb15, Jannifer Oliveira Chiang15, Edson Delatorre12#, Gonzalo Bello2#.
#These senior authors contributed equally to this article.
1. Núcleo de Vigilância de Vírus Emergentes, Reemergentes ou Negligenciados – ViVER/EDTA. Instituto Leônidas e Maria Deane, Fiocruz, Manaus, AM, Brazil;
2. Laboratório de Arbovírus e Vírus Hemorrágicos, Instituto Oswaldo Cruz, Fiocruz, Rio de Janeiro, RJ, Brazil;
3. Diretoria de Ensino e Pesquisa, Fundação Centro de Controle de Oncologia do Estado do Amazonas, FCecon, Manaus, AM, Brazil
4. Laboratório Central de Saúde Pública de Roraima, Boa Vista, RR, Brazil;
5. Fundação de Vigilância em Saúde – Dra. Rosemary Costa Pinto, Manaus, AM, Brazil;
6. Laboratório Central de Saúde Pública de Rondônia, Porto Velho, RO, Brazil;
7. Laboratório Central de Saúde Pública do Acre, Rio Branco, AC, Brazil;
8. Núcleo de Doenças de Transmissão Vetorial, Secretaria Estadual de Saúde do Acre, Rio Branco, AC, Brazil;
9. Coordenação Geral de Laboratórios de Saúde Pública – CGLAB, Ministério da Saúde, Brasília, DF, Brazil;
10. Laboratório de Virologia Molecular, Fiocruz Rondônia, Porto Velho, RO, Brazil;
11. Gerência de Virologia, Fundação de Medicina Tropical – Dr. Heitor Vieira Dourado, Manaus, AM, Brazil;
12. Laboratório de Genômica e Ecologia Viral, Departamento de Patologia, Centro de Ciências da Saúde, Universidade Federal do Espírito Santo, Vitória, ES, Brazil
13. Laboratório de Virologia Molecular, Instituto Carlos Chagas, Fiocruz, Curitiba, PR, Brazil
14. Instituto Aggeu Magalhães, Fiocruz, Recife, PE, Brazil
15. Department of Arbovirology and Hemorrhagic Fevers, Evandro Chagas Institute, Health and Environment Surveillance Secretariat, Ministry of Health, Ananindeua, PA, Brazil.
Main text
Oropouche virus (OROV) is an arthropod-borne pathogen of the genus Orthobunyavirus, family Peribunyaviridae, Orthobunyavirus oropoucheense species. This virus is maintained in nature in a sylvatic cycle that still needs to be well defined. However, there is evidence of the participation of forest mosquitoes like Aedes serratus and Coquillettidia venezuelensis and mammals such as sloths and non-human primates as key reservoirs. On the other hand, biting midges (Culicoides paraensis) are considered the primary vector of the urban cycle, although Culex quinquefasciatus mosquitoes have already been implicated in OROV transmission (1). In the last 70 years, at least 30 human outbreaks of OROV have been reported in Latin American countries (Brazil, Peru, Colombia, French Guiana, and Panama). Due to the recurrent resurgence of OROV in human populations of the Amazon region and the notable increase in the incidence and geographical spread of reported OROV infections in recent years, it is one of the most significant emerging arboviral threats in Latin America (2).
Until the emergence of chikungunya (CHIKV) and Zika (ZIKV) viruses between 2014-2015, OROV was the arbovirus with the highest incidence in Brazil, only surpassed by dengue virus (DENV) (3). The country’s largest documented OROV outbreak was reported in the late 1970s, where estimates point to more than 100,000 human cases (4), but the true burden of OROV-induced disease remains unknown because direct measurements of the human population incidence were not performed and has been rarely conducted during other outbreaks and inter epidemic periods due to limited or no systematic surveillance. Moreover, despite an extended history of outbreaks, the number of full-length OROV genomes obtained in Brazil since its first identification in 1960 is scant (n = 44) and mostly restricted to only two states from the eastern Amazonian region (Pará and Amapa), making difficult to understand the evolutionary and dissemination dynamics of this arbovirus (5). Thus, there is a crucial need to expand the diagnostic and sequencing efforts to enhance our comprehension of the molecular evolution, transmission dynamics and the disease burden of OROV in the Amazon region.
Here, we report epidemiological and molecular/genomic evidence of large-scale OROV outbreaks occurring in four Brazilian states of the western Amazon region between 2022 and 2024: Amazonas (AM), Acre (AC), Rondônia (RO) and Roraima (RR). Almost 1,000 individuals tested positive for OROV and 75 new full-length OROV genomes were generated from those recent outbreaks in the Brazilian Amazon region. Our findings revealed that recent Brazilian outbreaks resulted from the sustained transmission and dissemination of a novel OROV reassortant lineage.
Between January 2022 and January 2024, 946 individuals presenting symptoms consistent with arbovirus infection in the Brazilian states of AM, AC, RO and RR, tested positive for OROV (OROV+) in a real-time RT-PCR protocol (6) currently in use in some Brazilian states. The distribution of OROV+ cases during this period was as follows: 642 cases in AM, 148 cases in RR, 118 cases in AC and 38 cases in RO (Fig. 1a). Focusing specifically on AM state, where the most extensive sampling occurred, OROV was the second most frequently detected arbovirus, outmatched only by DENV-2 (829 cases). A small first wave of OROV cases was registered between late 2022 and early 2023, followed by a second, more significant wave starting in October 2023 (EW41) that dominated the epidemiological landscape of arboviral infections during December 2023 and January 2024 (Fig. 1b). Interestingly, the current OROV outbreak predominantly affects males (55% of the cases), presenting a distinct gender profile compared to other co-circulating arboviruses (DENV, CHIKV, and ZIKV) that mostly affected females (55% of the cases). About half of OROV+ cases in AM and AC were detected in the age group of 25–44 years (Fig. 1c). The adjustment of ages in the studied population to a Gaussian model revealed a slight deviation towards older ages in the population with OROV+ diagnosis (35.2 ± 6.4 years) concerning the populations with DENV+ diagnosis (32.4 ± 17.6 years) and arbovirus negative diagnosis (29.4 ± 19.4 years) (Fig. 1c).
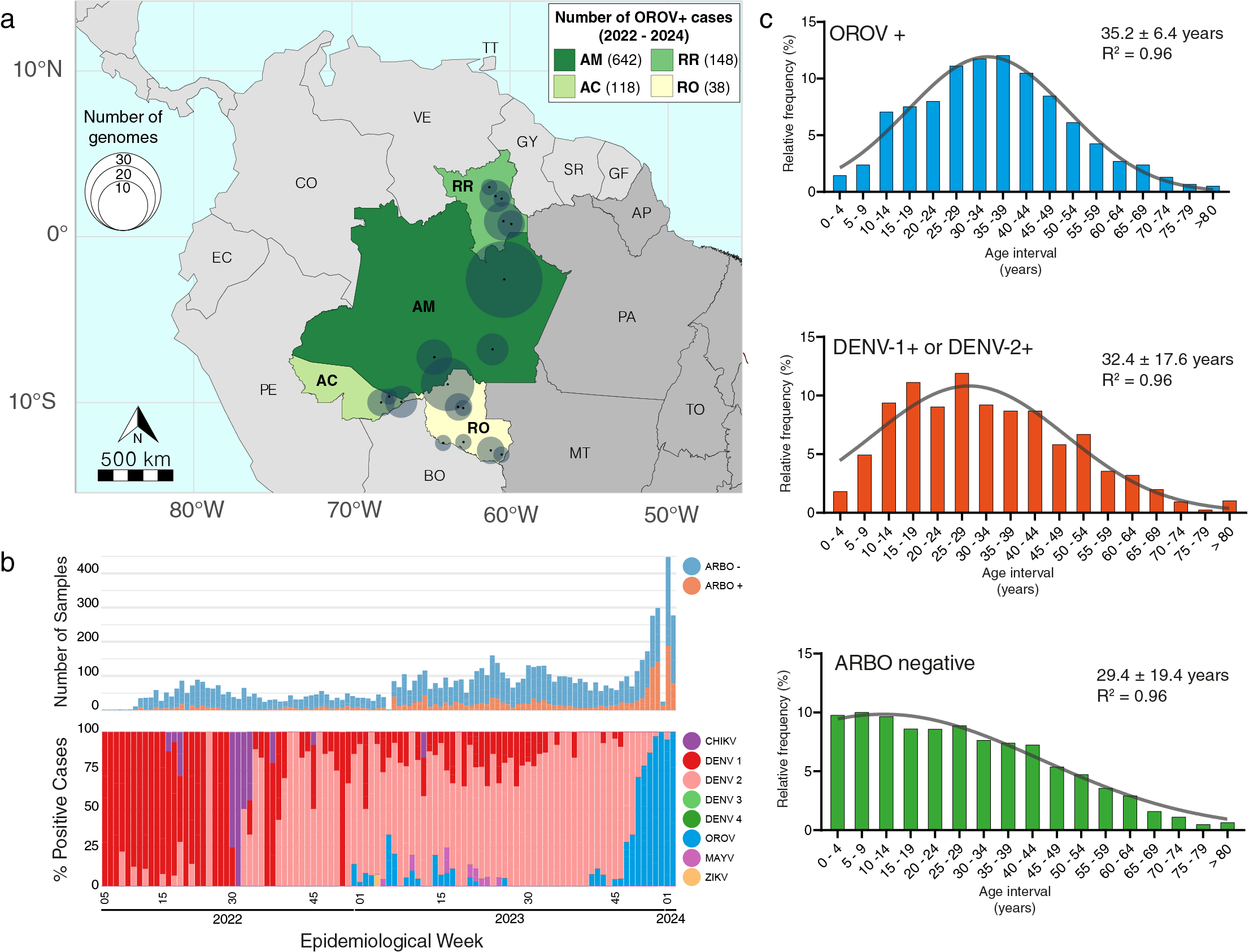
Figure 1. Epidemiological characteristics of the OROV outbreak in the western Amazonian Brazilian region - 2022-2024. a) Geographic distribution of the OROV+ cases diagnosed in Brazil between January 2022 and January 2024. State colors represent the number of confirmed OROV+ cases, and circle sizes indicate the number of genomes per municipality. AC - Acre, AM - Amazonas, RO - Rondônia, RR - Roraima, PA - Pará, AP - Amapá, MT - Mato Grosso, TO - Tocantins. b) Number of samples analyzed by real-time RT-PCR for arboviruses per epidemiological week (top panel) and molecular positivity for arboviruses in the AM state, Brazil, from January 2022 to January 2024 (bottom panel). c) Histograms depicting the age distribution of the study patients in AM plus AC with different arboviral diagnosis profiles. Samples classified as ARBO-negative tested negative for DENV, ZIKV, CHIKV, OROV and Mayaro virus (MAYV).
To understand the origin and spread of the OROV driving recent outbreaks in the Brazilian Amazon region, we sequenced full-length OROV genomes with an amplicon-based strategy designed by our group using Primal Scheme (7), a modified version of Primer3 (8) embedded in Geneious Prime 2023.2.1, and Illumina COVIDSeq as the backbone for a more cost-effective genome-wide sequencing strategy for a larger set of samples (OROV Primers sequences available upon request). We sequenced full-length OROV genomes from 75 patients living in 18 cities from the Brazilian states of AM (n = 37), RO (n = 20), RR (n = 12), and AC (n = 6), collected between 30th August 2022 and 12th December 2023 (Fig. 1a). The Central Laboratories from the States of AM (LACEN-AM), RR (LACEN-RR), RO (LACEN-RO), and AC (LACEN-AC) sent OROV positive samples for sequencing at Instituto Leônidas e Maria Deane (ILMD - FIOCRUZ Amazônia), that is the hub of FIOCRUZ Genomics Surveillance Network of the Brazilian Ministry of Health in Manaus, AM. The epidemiological information regarding date and place of collection for all samples analyzed in this study and their phylogenetic context (considering the M segment) may be visualized in detail on our Microreact (9) page: https://microreact.org/project/fn-orov-2022-2023. The 75 complete OROV sequences of S, M, and L genomic segments generated in this study were aligned with corresponding segments of all published full-length OROV genome sequences available at the NCBI (n = 72), sampled in the Americas between 1955 and 2021, and with the prototype sequences of OROV (OROVP L: AF484424; M: AF441119; S: AY237111), Iquito virus (IQTVP L: KF697142; M: KF697143; S: KF697144), Perdoes virus (PEDVP L: KP691627; M: KP691628; S: KP691629), and Madre de Dios virus (MDDVP L: KF697147; M: KF697145; S: KF697146). All these viruses are classified in the species Orthobunyavirus oropoucheense, having the classical OROV as the exemplar isolate of the species (Genus: Orthobunyavirus | ICTV). The resulting datasets were used to infer phylogenetic trees using the Bayesian statistical framework of MrBayes 3.2 (10).
From the Bayesian phylogenetic tree we identified two well-supported (PP > 0.75) major viral lineages for the M (M1 and M2) and L (L1 and L2) segments and three major viral lineages for the S segment (S1, S2 and S3) (Fig. 2a-c). OROV sequences from Brazil (2022-2023) branched in a highly supported (PP > 0.90) monophyletic clade (OROVBR-2015-2023) across all three genomic segments together with an OROV genome from French Guiana sampled in 2020 (11) and another OROV genome obtained from Tefé, AM, Brazil, in 2015 (12) (Fig. 2a-c). For the M segment, the OROVBR-2015-2023 clade branched within the M1 lineage that comprises the OROVP prototype and was more closely related to a sister monophyletic clade (OROVBR-2009-2018), comprising sequences detected in the eastern Amazonian states of Amapá (AP) in 2009 and Pará (PA ) in 2018 (Fig. 2a). In the L and S segments, the OROVBR-2015-2023 clade branched within the L2 and S2 lineages, which include the IQTVP prototype. This clade is nested within a basal paraphyletic clade (OROVPE/CO/EC-2008-2021), encompassing sequences sampled in Peru, Colombia, and Ecuador between 2008 and 2021 (Figs. 2b-c). Interestingly, the OROVBR-2009-2018 clade branched with high support with the PEDVP prototype in both L (L2 lineage) and S (S3 lineage) segments. In contrast, the OROVPE/CO/EC-2008-2021 clade branched together with sequences detected in Panama and Peru between 1989 and 2000 in the M segment (M2 lineage lineage), separated from the OROV prototype and sequences sampled in Brazil between 1960 and 2023 (Fig. 2a-c).
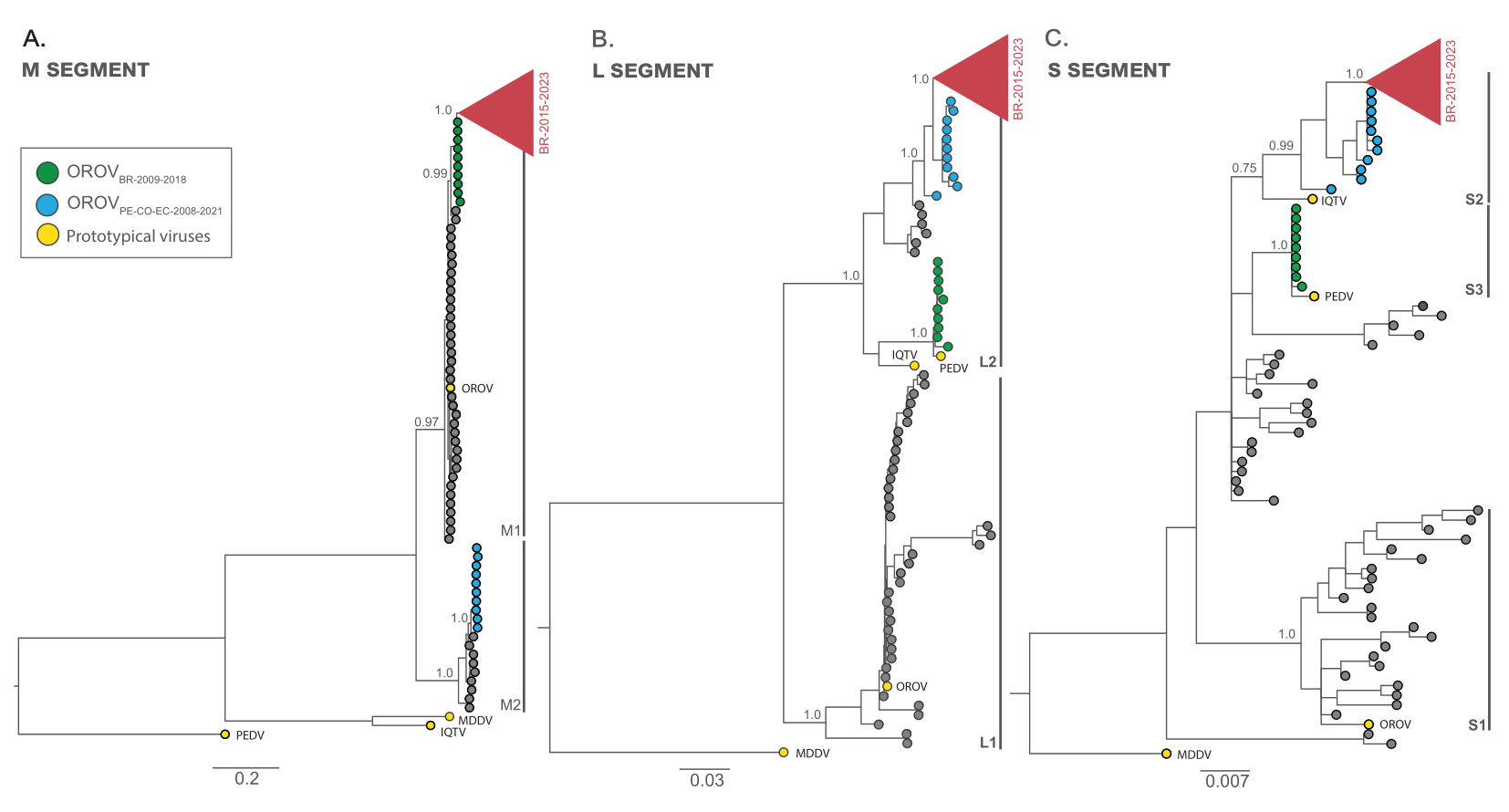
Figure 2. Majority-rule consensus Bayesian phylogenetic trees inferred from the OROV datasets of M, L and S. The phylogenetic trees depict the evolutionary history of OROV segments M (n = 147) (a), L (n = 147) (b) and S (n = 146) (c). The tips of prototypical viruses and major clusters are color-coded according to the legend in the upper right corner. Gray brackets are employed to demarcate major monophyletic lineages. The trees include annotations indicating the posterior probability support for both major lineages and specific clades. All trees are drawn in accordance to the genetic distance scale in the bottom of each panel. IQTV: Iquitos virus, MDDV: Madre de Dios virus, OROV: Oropouche virus, PEDV: Perdões virus.
The phylogenetic discordance between genomic segments is consistent with the occurrence of successive reassortment events throughout the evolutionary timeline of OROV in South America, as schematically depicted in Fig. 3. According to this hypothesis, the clade OROVBR-2015-2023 that is currently spreading in Brazil was identified as an M1L2S2 reassortant virus that acquired the M segment from the clade OROVBR-2009-2018 (M1L2S3) and the L+S segments from the clade OROVPE/CO/EC-2008-2021 (M2L2S2), which has been circulating in the Amazon basin since the late 2000s.
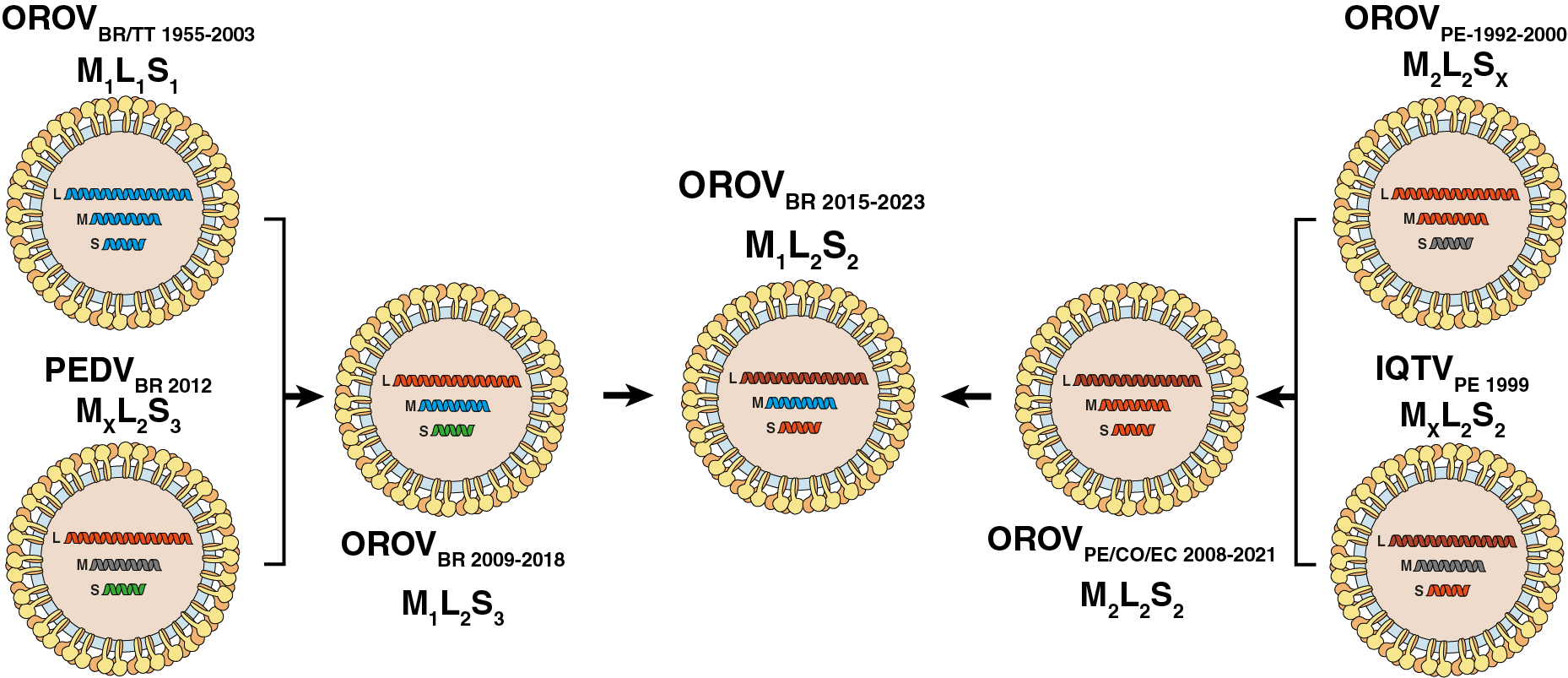
Figure 3. Putative reassortment events that generated the current genomic diversity of OROV in South America. Each OROV genomic segment is colored according with the lineage identified in this study. Illustrations were obtained from SwissBioPics (https://viralzone.expasy.org/, (13)) and modified to align with the reassortment events hypothesis. Oropouche virus (OROV), Iquito virus (IQTV), Perdoes virus (PEDV).
The clade OROVBR-2015-2023 and their most closely related sequences of the clades OROVPE/CO/EC-2008-2021 (L and S segments) and OROVBR-2009-2018 (M segment) were further analyzed to estimate the time of the most recent common ancestor (TMRCA) and the time interval of the reassortment event that originated the recent clade. The temporal structure of the selected sequences was assessed using the program TempEst v.1.5.3 (14) by a root-to-tip linear regression, in which the significance of the collection date/genetic distance correlation was accessed with a Spearman correlation test. Time-scaled phylogenetic trees were estimated using the non-parametric Bayesian skyline coalescent demographic model and a relaxed molecular clock model implemented in the software BEAST 1.10 (15). The correlation between genetic divergence and sampling time was significant for datasets of all three genomic segments (p < 0.05). However, the temporal structure of the shortest S segment (R2 = 0.56) was weaker than that of the L (R2 = 0.77) and M (R2 = 0.81) segments (Fig. 4a-c). The median estimated evolutionary rate for all three segments was roughly similar and ranged between 0.9 - 1.3 × 10-3 substitutions/site/year (Fig. 4a-c). Analyses of all three genomic segments traced back the median TMRCA of the clade OROVBR-2015-2023 to 2013-2014, short before the sampling time of the oldest sequence from that clade detected in Tefé-AM in 2015. The time frame of the reassortment event between clades OROVBR-2009-2018 and OROVPE/CO/EC-2008-2021 was estimated between 2008 and 2013 (Fig. 4d-f).
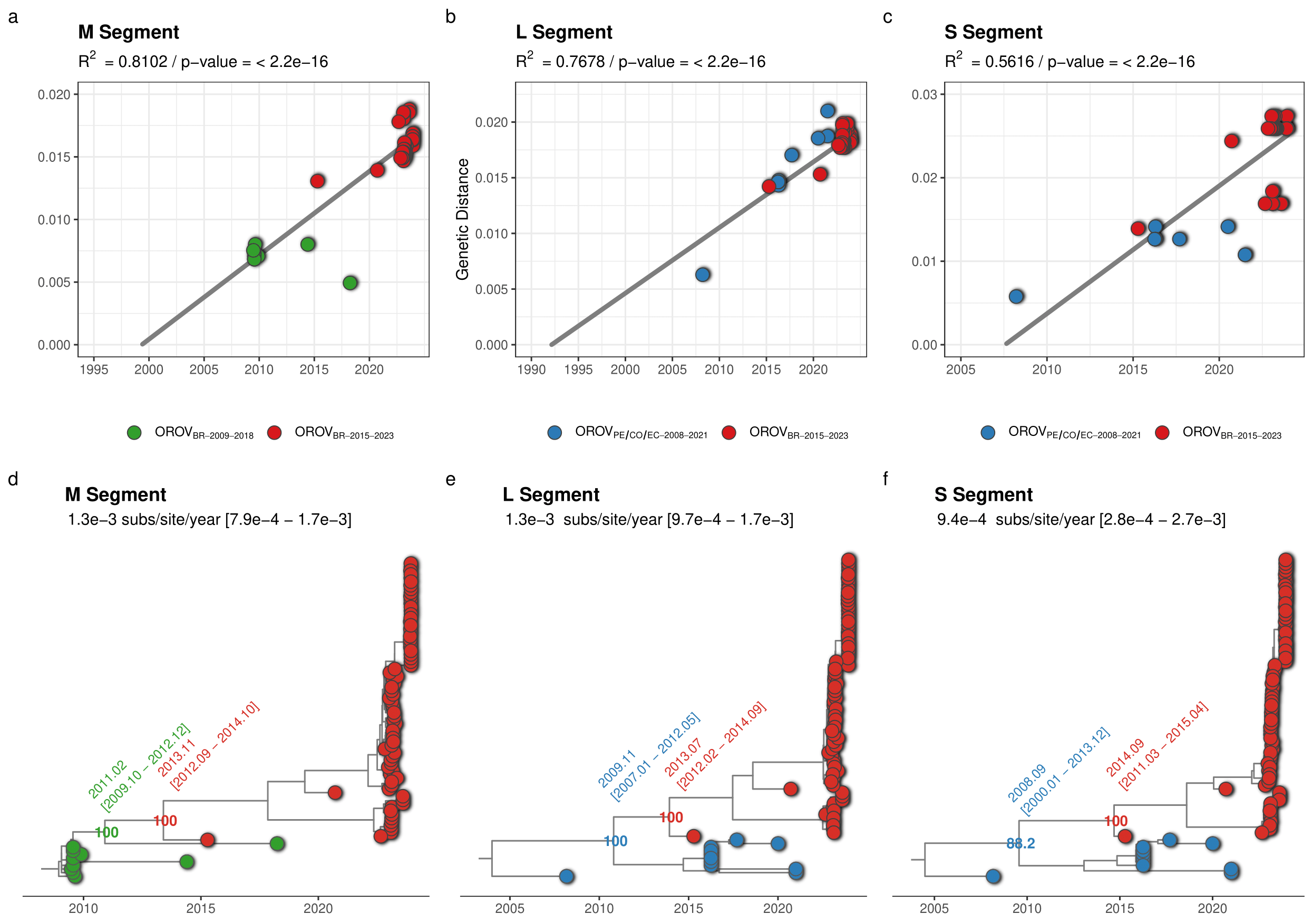
Figure 4. Temporal dynamics of the current outbreak OROVBR-2015-2023 clade and the most closely related clades. The figure represents the root-to-tip linear regression between collection date and genetic distance (a-c) and time-scaled Bayesian phylogenetic trees (d-f) inferred with datasets composed of the OROVBR-2015-2023 clade (red tips) and its most closely related sequences of the OROVPE/CO/EC-2008-2021 clade (blue tips) in the L (n = 88) and S (n = 88) segments and of the OROVBR-2009-2018 clade (green tips) in the M segment (n = 87). The molecular clock rate inferred for each segment is annotated in each tree, as are the posterior probability support and the median TMRCA (alongside its 95% HPD interval) of key nodes.
These findings revealed that the year-long maintenance and spread of a novel reassortant OROV lineage in the Brazilian Amazon region was associated with the recent upsurge of OROV cases in large urban centers like Manaus. This finding combined with the recent identification of imported OROV+ cases in São Paulo (one patient arriving from AM) and Curitiba (two patients, one from AM e other from AC) (unpublished data), rise concerns about the risk of spreading and establishment of this neglected arbovirus into urban areas outside the Amazon region. Further studies are needed to assess the potential impact of recent reassortment events in OROV on vector competence, disease severity and virus transmissibility in susceptible populations from non-endemic regions. Our study also warns about the urgent need to implement sensitive and specific immunological and genetic diagnostic tests, like the one developed by our group (6), in all Brazilian states and perhaps other Latin American Countries, considering the clinical similarity between OROV infections and other common acute febrile arboviral illnesses endemic in Brazil caused by DENV, ZIKV, and CHIKV.
This report addresses an ongoing large-scale OROV outbreak that, at the time of writing, is still unfolding. Therefore, we will update this post and following manuscripts in light of more and new data as soon as the information is available and analyzed. We strive to provide as much data as possible in order to address and mitigate this outbreak of public health concern.
Acknowledgments
The authors wish to thank all the healthcare workers from the Brazilian States fighting the current Oropouche outbreak. We also appreciate the support of FIOCRUZ COVID-19 Genomics Surveillance Network members, the General Laboratory Coordination (CGLab) of the Brazilian Ministry of Health (MoH), Brazilian Central Laboratory States (LACENs) and health surveillance agencies from Acre, Amazonas, Rondônia and Roraima. Funding support FAPEAM Call 04/2022/FIOCRUZ/FAPEAM/FAPERO - INOVAÇÃO NA AMAZÔNIA; Amazônia +10; FAPEAM Call 023/2022 - INICIATIVA AMAZÔNIA +10; Rede Genômica de Vigilância em Saúde - REGESAM); Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq, e Fundação de Amparo à Pesquisa do Espírito Santo.
Data availability
All the OROV genomes generated and analyzed in this study were deposited at GenBank under accession numbers PP153945 to PP154172. Due to our commitment to public health, we asked for the immediate release of those genomes as soon as the GenBank staff processes them.
Bibliography
- Travassos da Rosa JF, de Souza WM, Pinheiro F de P, Figueiredo ML, Cardoso JF, Acrani GO, et al. Oropouche Virus: Clinical, Epidemiological, and Molecular Aspects of a Neglected Orthobunyavirus. Am J Trop Med Hyg. 2017 May;96(5):1019–30.
- Wesselmann KM, Postigo-Hidalgo I, Pezzi L, De Oliveira-Filho EF, Fischer C, De Lamballerie X, et al. Emergence of Oropouche fever in Latin America: a narrative review. Lancet Infect Dis. 2024 Jan;S1473309923007405.
- Figueiredo LTM. Emergent arboviruses in Brazil. Rev Soc Bras Med Trop. 2007;40(2):224–9.
- Sakkas H, Bozidis P, Franks A, Papadopoulou C. Oropouche Fever: A Review. Viruses. 2018 Apr 4;10(4):175.
- Gutierrez B, Wise EL, Pullan ST, Logue CH, Bowden TA, Escalera-Zamudio M, et al. Evolutionary Dynamics of Oropouche Virus in South America. Parrish CR, editor. J Virol. 2020 Feb 14;94(5):e01127-19.
- Naveca FG, Nascimento VA do, Souza VC de, Nunes BTD, Rodrigues DSG, Vasconcelos PF da C. Multiplexed reverse transcription real-time polymerase chain reaction for simultaneous detection of Mayaro, Oropouche, and Oropouche-like viruses. Mem Inst Oswaldo Cruz. 2017 Jul;112(7):510–3.
- Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc. 2017 Jun;12(6):1261–76.
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012 Aug;40(15):e115.
- Argimón S, Abudahab K, Goater RJE, Fedosejev A, Bhai J, Glasner C, et al. Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microb Genomics. 2016 Nov;2(11):e000093.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012 May;61(3):539–42.
- Gaillet M, Pichard C, Restrepo J, Lavergne A, Perez L, Enfissi A, et al. Outbreak of Oropouche Virus in French Guiana. Emerg Infect Dis. 2021 Oct;27(10):2711–4.
- Naveca FG, Nascimento VA, Souza VC, Figueiredo RMPD. Human Orthobunyavirus Infections, Tefé, Amazonas, Brazil. PLoS Curr [Internet]. 2018 [cited 2024 Jan 23]; Available from: Human Orthobunyavirus Infections, Tefé, Amazonas, Brazil – PLOS Currents Outbreaks
- Hulo C, de Castro E, Masson P, Bougueleret L, Bairoch A, Xenarios I, et al. ViralZone: a knowledge resource to understand virus diversity. Nucleic Acids Res. 2011 Jan;39(Database issue):D576-582.
- Rambaut A, Lam TT, Max Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016 Jan;2(1):vew007.
- Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018 Jan;4(1):vey016.