This topic is about a recently released genome sequence from a patient, infected in Monrovia, Liberia and then repatriated to the US. The sequence (Genbank accession KP178538) was submitted by Shannon Whitmer, C. Albarino. and Ute Stroeher of the CDC.
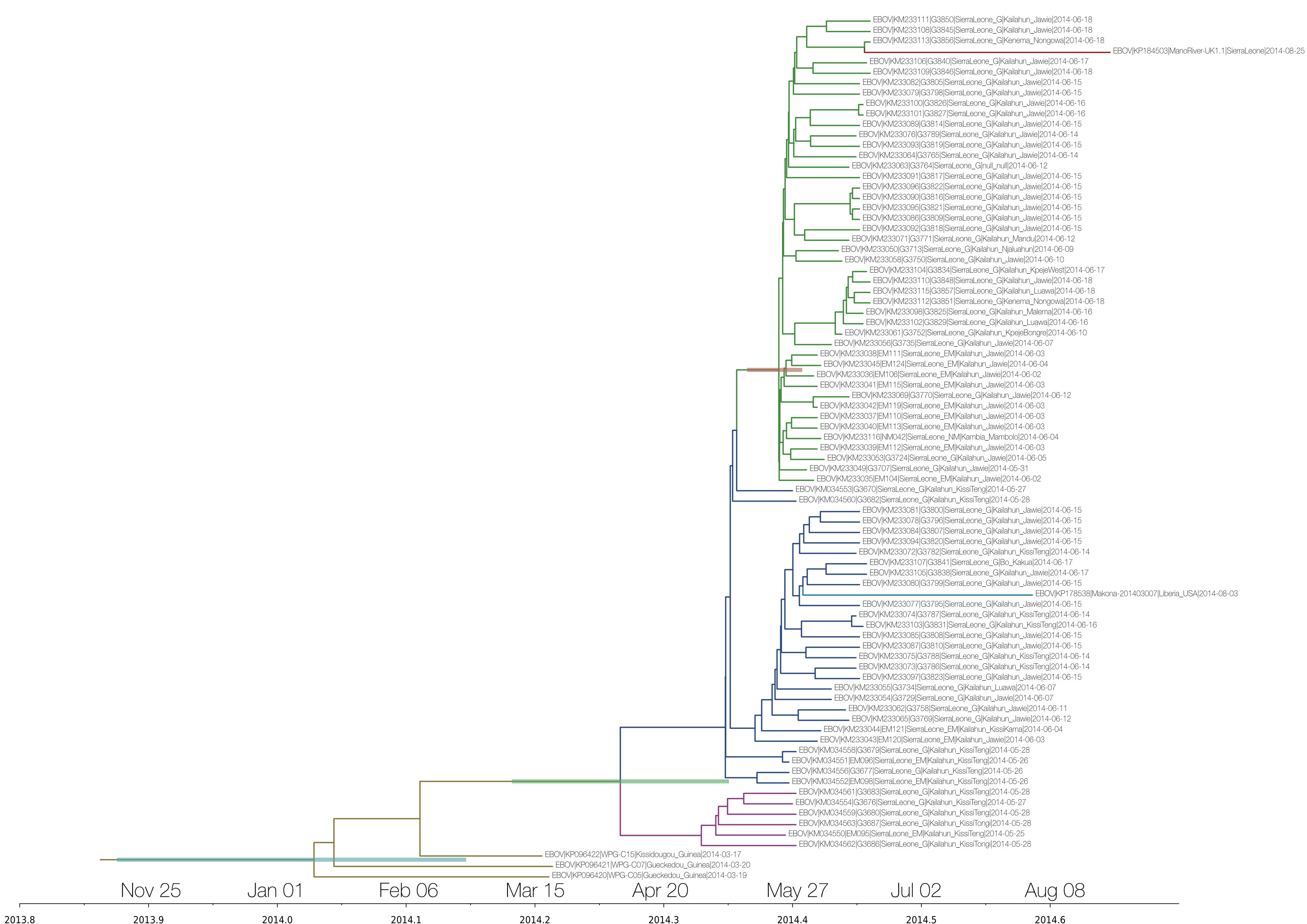
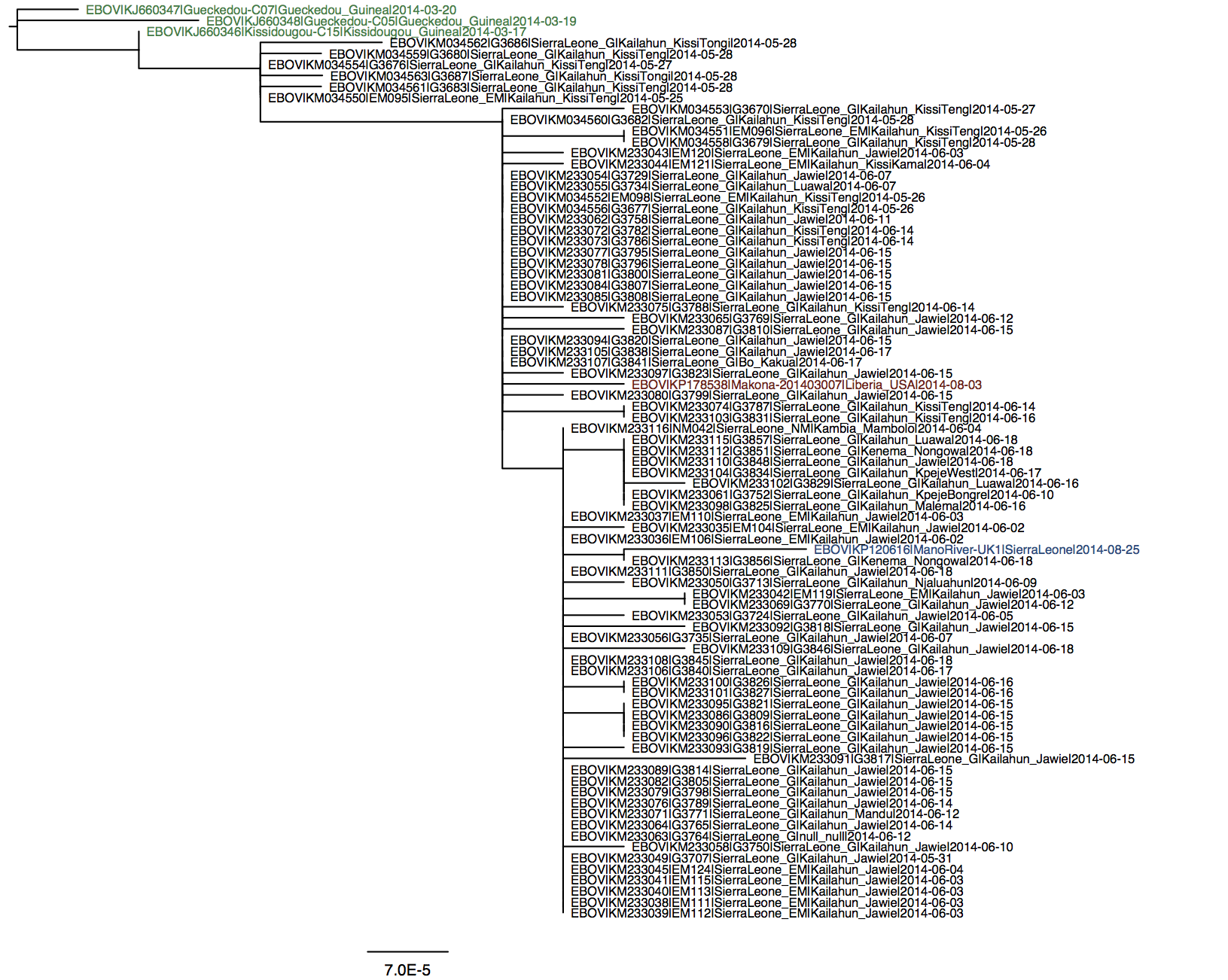
A simple maximum likelihood tree of the full genomes with available Guinea and Sierra Leone sequences suggest a relationship with Sierra Leone cases from May (it falls into ‘clade SL2’ of Gire et al 2014).
I see two SNPs that separate this LIB sequence from SL clade 2 (e.g. KM233077.1): one at 4655 and one at 7657 (these are coordinates relative to Kissidougou / KJ660346.2). @evogytis pointed out that the latter one appears to be a reversion to an ancestral allele seen in pre-2014 EBOV, which is odd.
Hi Daniel - we recently met while I was visiting Broad two weeks ago. I hope that you are doing well!
Yes, we used CLC Genomics and bwa/samtools to assemble this sequence using reference KM233053. Any ambiguous bases called with bwa/samtools were resolved using the consensus sequence made with CLC Genomics.
If you are concerned that this approach introduced bias into the data, then I would be happy to try a de novo assembly with the reads. You mentioned when we met that you might be willing to share your de novo assembly scripts in the future… If this is still the case, then I can give those scripts a try with the reads for this sequence. Just let me know! Thanks!
Hi Shannon, yeah I’m curious because it looks very close to the Sierra Leone sequences, which is surprising, given that Liberian EBOV was not thought to have come from Sierra Leone. This is the first publicly released EBOV genome from Liberia (with the exception of KM251803, the small PCR fragment of the L gene from Nigeria ex Liberia). So before jumping to conclusions about country to country transmissions and rates of viral evolution, the question that comes to mind is how much the choice of reference might have influenced the final assembly, so it’d be helpful to see the raw read alignments against its own consensus. Are you up for sharing a BAM file?
I can work with you to try de novo assembly, but there’s a bunch of rough edges in our pipeline that make it harder to use (currently) outside our compute environment. I’m thinking within a few months, we’ll have those smoothed out. I can walk you through what is currently available (it’s quite messy at the moment), but if you’re okay with sharing reads, it may be faster if I give it a run myself and send you the resulting fastas and bams. You have my email address from your earlier visit, right?
Hi Shannon, it is amazing that you all released this genomic sequence. Thank you so much for doing this for the community. As Danny wrote, we would be happy to run the assembly for you, and would love to get you up and running on our pipeline when we have it available to run outside our evironment. As you are now part of the Broad family we would be happy also to work to get your a Broad collaborator computer account.
This is the “within-outbreak” tree run under UCLN, SRD06 and skygrid for 50M states and includes the new Liberian and Sierra Leonean sequences as well as the re-sequenced Guinea isolates. Presumably the extra SNPs in the latter push the root a bit deeper and now we’re getting into beginning of December 2013. The rate estimate is 1.3575E-3 subs/site/year and 95% HPDs are (7.9637E-4, 1.9384E-3), which is lower than what we’ve seen before and more similar to the “between-outbreak” tree, although it’s still higher, as expected.
@evogytis, I’m not sure what I think about using the new re-sequenced Guinea isolates… my understanding (@Kristian_Andersen correct me if I’m wrong) is that Heinz Feldmann’s group took the original three Baize samples through culture and then sequenced the result. (note the H.sapiens-tc name in Genbank), so it’s possible that some of the differences you see from the Baize sequences could be due to passaging in vitro. That would also push back your tMRCA.
Yeah, @dpark is correct - those additional SNPs in the resequenced could likely be due to passage-induced mutations. I don’t know how many times the virus was passaged, but those SNPs don’t really ‘match up’ with previous strains. @evogytis - have you tried rerunning this with the corrected GIN sequences that Baize/Günther sequenced? (I think they likely resequenced their own samples).
You’re right - GenBank says Vero E6 cells. I’ll re-run the analyses using the current version of original Guinea sequences (KJ660346, etc.). Or should I switch to N’d out sequences from the Science paper? By the way - as @rambaut mentioned there were some odd SNPs in the ManoRiver (repatriated UK) sequence so I’ve switched to using KP184503 (rather than KP120616), which contains two ambiguities towards the end of the genome, which I assume means that nothing was imputed.
Yeah for Guinea, we noticed that the .2 version of the Guinea sequences (KJ660346.2, etc) mostly differed from their .1 originals in the areas that we had N’ed out earlier, so it looks to be corrected. They are less distant from our SL sequences than in our original analysis, so I bet the tMRCA actually comes forward a little.
KP184503 is, again, a H.sapiens-tc sample, so it’s possible that this sequence may have been influenced by passaging in culture.
 we would be happy also to work to get your a Broad collaborator computer account.
we would be happy also to work to get your a Broad collaborator computer account.