Authors: Crystal M. Gigante, Jiusheng Deng, Hui Zhao, Victoria A. Olson, Todd G. Smith, Yu Li
Affiliation: Poxvirus and Rabies Branch, National Center for Emerging and Zoonotic Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, GA, USA
Summary: The current sequence for clade I monkeypox virus RefSeq (strain Zaire-96-I-16, accession NC_003310.1) contains many unique single nucleotide polymorphisms not seen in other clade I monkeypox virus sequences. Re-sequencing an early passage of Zaire-96-I-16 using current short read sequencing methods revealed 142 sequence differences relative to the published sequence.
In 2001, Shchelkunov et al. published the genome sequence of monkeypox virus (MPXV) strain Zaire-96-I-161. This genome has since been commonly used as a reference, since it contains many single nucleotide polymorphisms not seen in other MPXV genomes, and is currently listed as the RefSeq for MPXV clade I (RefSeq: NCBI Reference Sequence Database). With expanding sequencing capacity in endemic countries and recent mpox outbreaks, there has been rapid expansion in available MPXV genome sequences2, 3, 4. Comparison with the now over 1,000 clade I MPXV genomes available (gisaid.org, Home - Nucleotide - NCBI) reveal clusters of SNPs in the Zaire-96-I-16 strain sequence that are not found in any extant MPXV. Moreover, phylogenetic analysis places Zaire-96-I-16 on a long branch in a clade with extant MPXV (Figure 1A) not in a sister branch, as would be expected if the mutations in Zaire-96-I-16 were inherited from a common ancestor. This prompted us to re-sequence Zaire-96-I-16 from the CDC Poxvirus repository.
We performed direct metagenomic sequencing5 of MPXV strain Zaire-96-I-16 (passage 5) and MPXV strain Congo_2003_358 (previously sequenced in 2005 using primer walking and Sanger sequencing: DQ011154.16). Briefly, virus was propagated in BSC-40 cells, infected cells were collected by centrifugation and lysed by several freeze thaw cycles. Total DNA extraction was performed on 100 µL crude virus using a Qiagen EZ1/2 DNA Tissue extraction kit after sample inactivation at 56°C for 15 minutes with Qiagen AL buffer and proteinase K at a 1:1:0.1 ratio. 15 µL of total DNA was used as input for DNA sequencing on an Illumina NovaSeq 6000 after library preparation using the Illumina DNA Prep Kit with half reagent volumes throughout5. Raw fastq files were used as input into the polkapox pipeline (GitHub - CDCgov/polkapox) for read filtering and de novo assembly with unicycler. Final genome sequences were generated by manual assembly of unicycler contigs using a reference-based consensus sequence generated using ivar version 1.3.1 (ivar consensus -q 20 -t 0.67 -m 10) using references (NC_003310.1 or DQ011154.1). Annotation and submission to NCBI databases was performed using TOSTADAS (GitHub - CDCgov/tostadas: 🧬 💻 TOSTADAS → Toolkit for Open Sequence Triage, Annotation and DAtabase Submission). Genome alignments were performed using mafft v.7.490 in Geneious prime 2024.0.7 (GraphPad Software), and phylogenetic analysis was performed using iqtree v.2.2.6 using model finder (-m MFP) with 1000 bootstrap replicates (-B 1000).
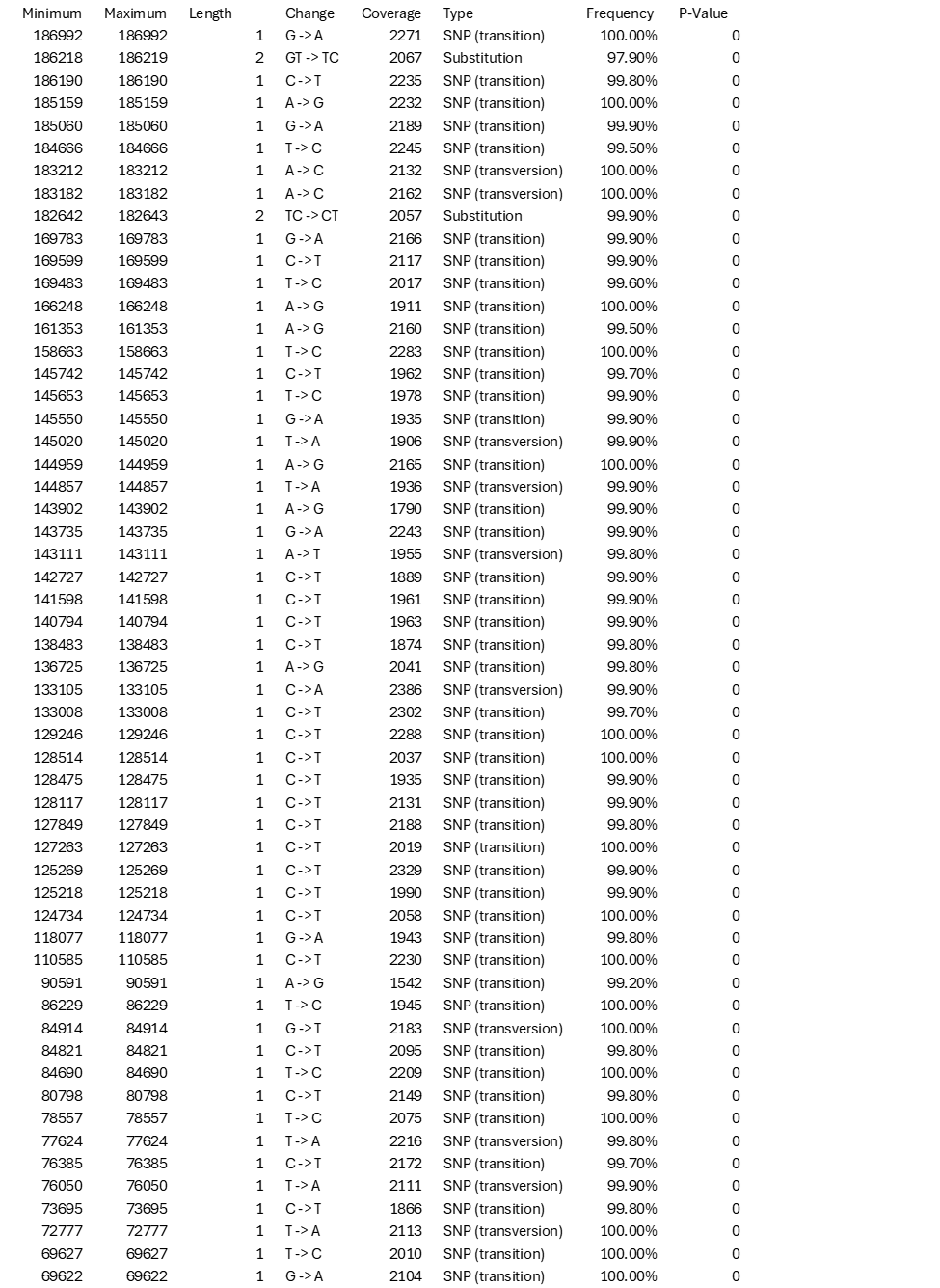
The resulting coding compete genome was 196,981 bp, with 33.1% GC content. The new sequence had 142 sequence changes relative to NC_003310.1 (Table 1); no differences were found for strain Congo_2003_358. Phylogenetic analysis placed the original Zaire-96-I-16 on a long branch, while the new genome fell on a shorter branch with other sequences from Democratic Republic of Congo (Figure 1). This new genome is available via GenBank Accession PX667572. Our re-sequencing supports that the unique, strain-specific SNPs in NC_003310.1 may be the result of sequencing errors in the original submission. The sequence differences are likely due to improved accuracy of current sequencing technologies compared to the original approach using a mix of plasmid subcloning/sequencing by Maxam-Gilbert technique and primer-walking/sequencing by Sanger sequencing1. We propose using this new genome in place of NC_003310.1 and replacement or update of the RefSeq.
Data availability: Data has been deposited in NCBI databases under the GenBank PX667572; BioSample: SAMN53797122; SRA: SRS27433776.
Acknowledgments: We thank members of the Poxvirus and Rabies Branch. The findings and conclusions in this report are those of the author(s) and do not necessarily represent the official position of the United States Centers for Disease Control and Prevention.

Figure 1. Phylogenetic tree of MPXV strain Zaire-96-I-16 with 22 clade Ia MPXV genomes. A. Original placement of sequence NC_003310.1 is shown in red. B. Updated sequence is shown in blue. Congo 2003-358 sequence is shown in pink to demonstrate no change in phylogenetic placement of re-sequenced isolate. Scale bar is in substitutions per site; support values are shown on nodes as percentages based on 1000 bootstraps. Support values are not shown on terminal nodes for ease of viewing, but all were >74%.
Table 1. Details of polymorphisms identified in newly generated sequences for MPXV strain Zaire-96-I-16 relative to NC_003310.1. Changes in tandem repeats, deletions or insertions were not considered.
References
1. Shchelkunov SN*, et al.* Human monkeypox and smallpox viruses: genomic comparison. FEBS Lett 509, 66-70 (2001).
2. Kinganda-Lusamaki E*, et al.* Clade I mpox virus genomic diversity in the Democratic Republic of the Congo, 2018-2024: Predominance of zoonotic transmission. Cell 188, 4-14 e16 (2025).
3. Parker E*, et al.* Genomics reveals zoonotic and sustained human mpox spread in West Africa. Nature 643, 1343-1351 (2025).
4. Ndodo N*, et al.* Distinct monkeypox virus lineages co-circulating in humans before 2022. Nat Med 29, 2317-2324 (2023).
5. Gigante CM, Weigand MR, Li Y. Orthopoxvirus Genome Sequencing, Assembly, and Analysis. In: Vaccinia, Mpox, and Other Poxviruses: Methods and Protocols). Springer US (2024).
6. Likos AM*, et al.* A tale of two clades: monkeypox viruses. J Gen Virol 86, 2661-2672 (2005).