Posted on behalf of the Swiss National Reference Center for Emerging Viral Infections, Geneva University Hospitals and the Institute of Medical Virology, University of Zurich (Partner Laboratory National Reference Center for Emerging Viral Infections).
On 5 May 2026, the Swiss National Reference Center for Emerging Viral Infections (Geneva University Hospitals) confirmed a case of Andes strain in a Swiss resident who had travelled on the MV Hondius cruise ship. The virus was sequenced from blood samples jointly by the Institute of Medical Virology (University of Zurich) and the Swiss National Reference Center for Emerging Viral Infections (Geneva University Hospitals) using Illumina technology (MiSeq instrument). The consensus sequence was generated with a minimum coverage of 5 reads.
The complete ANDV/Switzerland/Hu-3337/2026 consensus sequence for each of the 3 segments can be found here:
ANDV-Switzerland-Hu-3337-2026.fasta.gz (3.8 KB)

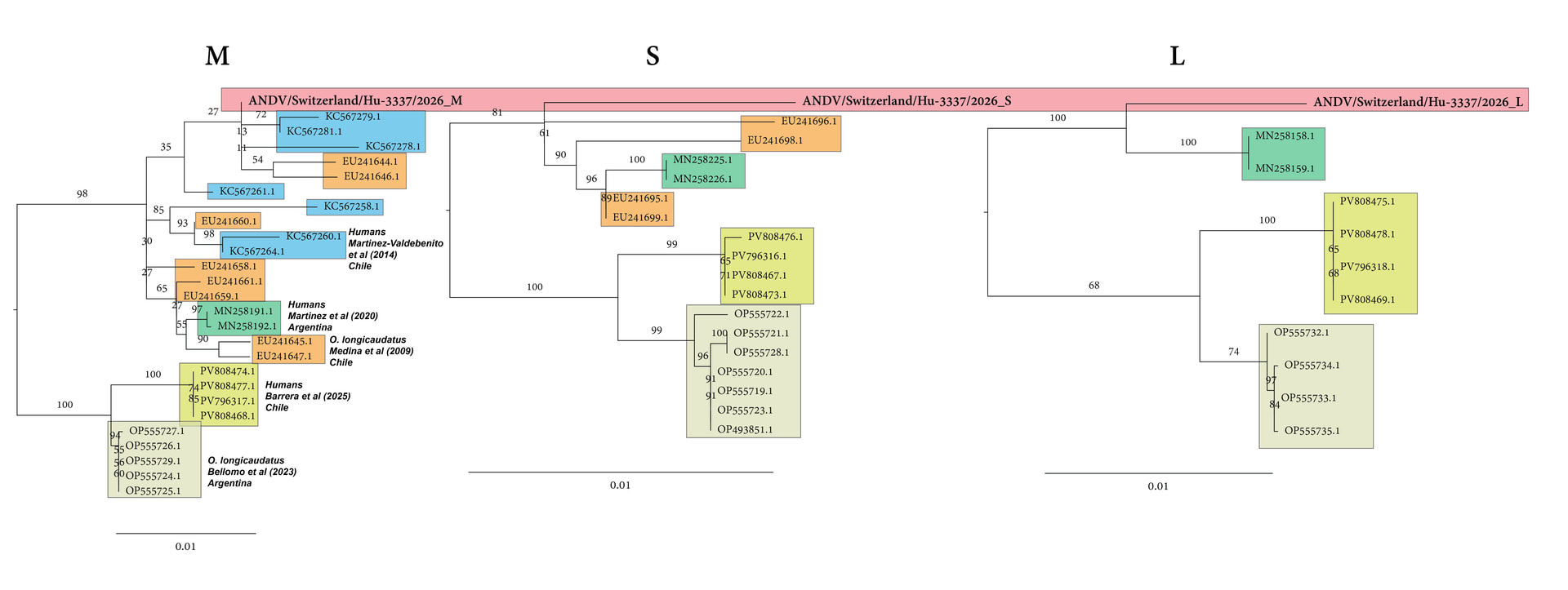
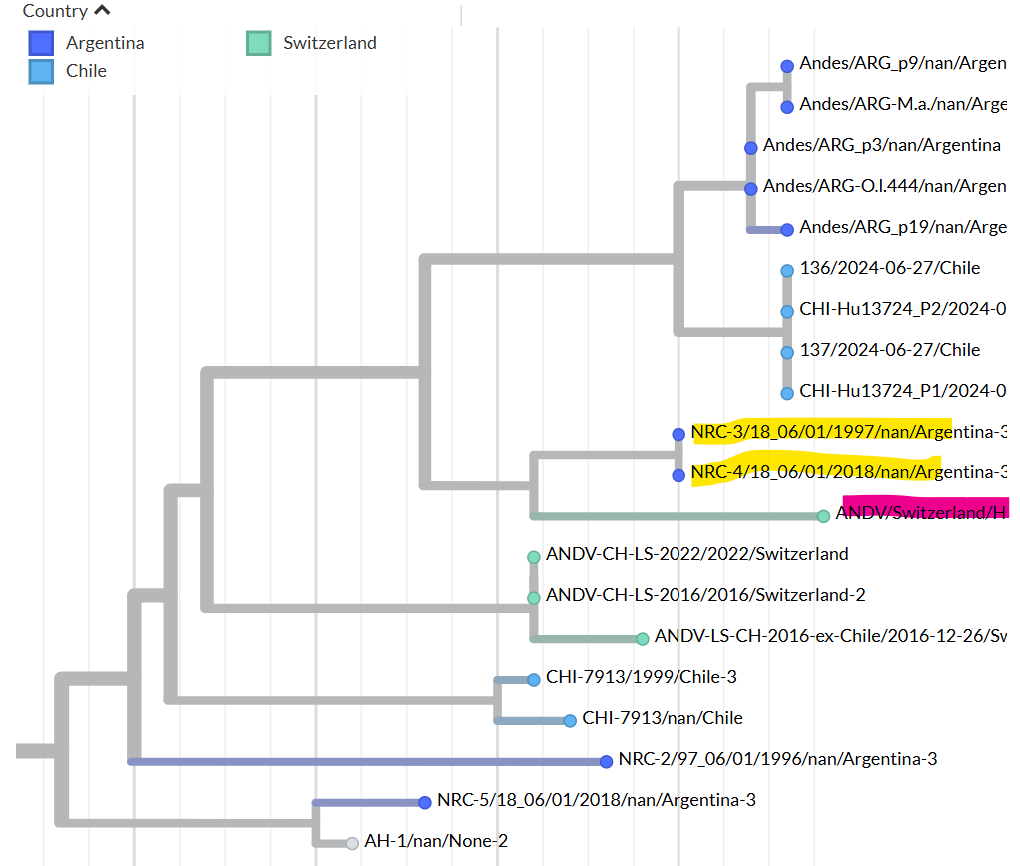
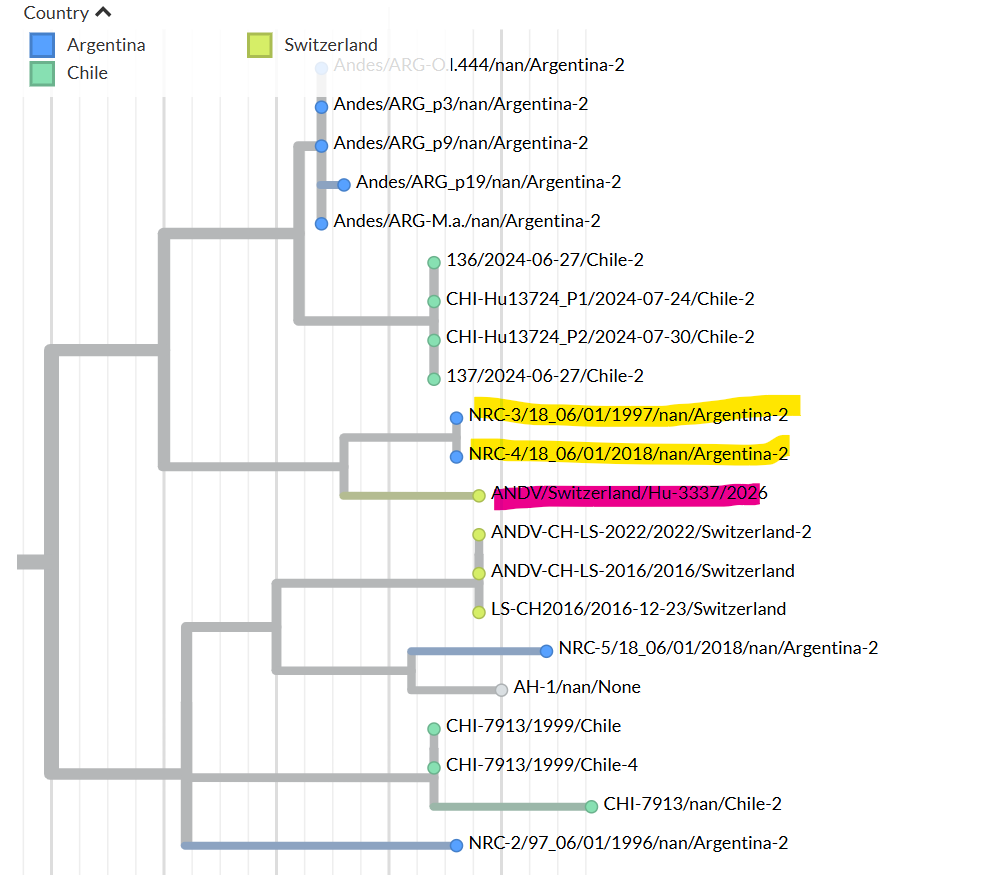
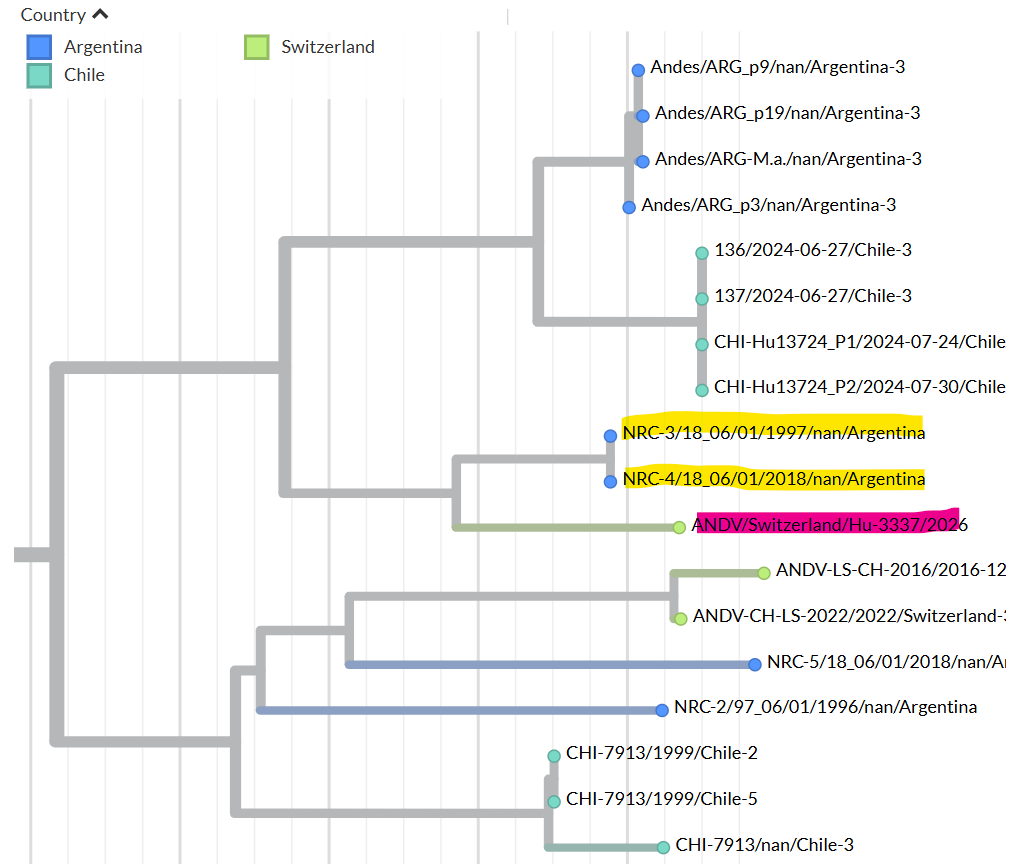
Figure 1: Maximum-likelihood tree based on the complete nucleocapsid coding sequence from the S segment of Andes virus strains.
Authors (in alphabetical order):
Bloemberg Guido - University of Zurich, Zurich, Switzerland
Chudzinski Valentin - Geneva University Hospitals, Geneva, Switzerland
Cordey Samuel - Geneva University Hospitals, Geneva, Switzerland
Huber Michael - University of Zurich, Zurich, Switzerland
Laubscher Florian - Geneva University Hospitals, Geneva, Switzerland
Pérez-Rodríguez Francisco-Javier - Geneva University Hospitals, Geneva, Switzerland
Pichler Ian - University of Zurich, Zurich, Switzerland
Schibler Manuel - Geneva University Hospitals, Geneva, Switzerland
Thomasson Valentine - Geneva University Hospitals, Geneva, Switzerland
Ziltener Gabriela - University of Zurich, Zurich, Switzerland