Preprint_CHIK_NGS_KDG_2023.pdf (276.4 KB)
Idrissa Dieng1*, Bacary Djilocalisse Sadio1*, Alioune Gaye2, Samba Niang Sagne3, Marie Henriette Dior Ndione1, Mouhamed Kane1, Mamadou Korka Diallo3, Bocar Sow3, Safietou Sankhe1, Amadou Diallo3, Madeleine Dieng1, Serge Freddy Moukaha Doukanda1, Maimouna Mbanne1, Seynabou Mbaye Ba Souna Diop1, Diamilatou Balde1, Mignane Ndiaye1, Khalidou Djibril Sow4, Maryam Diarra3, Abdoulaye Sam5, Ababacar Mbaye6, Boubacar Diallo7, Yoro Sall5, Ousmane Faye7, Boly Diop5, Abdourahmane Sow7, Amadou Alpha Sall1, Cheikh Loucoubar3, Ndongo Dia1, Oumar Faye1, Diawo Diallo2, Gamou Fall1, Scott C Weaver8, Mamadou Aliou Barry3, Mawlouth Diallo2, Moussa Moise Diagne1+
- Virology Department, Institut Pasteur de Dakar, Dakar 220, Senegal.
- Zoology Medical Department, Institut Pasteur de Dakar, Dakar 220, Senegal.
- Epidemiology, Clinical Research and Data Science Department, Institut Pasteur de Dakar, Dakar 220, Senegal.
- Health Emergency Operations Center, Ministry of Health, Dakar 220, Senegal.
- Prevention Department, Ministry of Health, Dakar 220, Senegal.
- Kedougou Medical Region, Ministry of Health, Kedougou 26005, Senegal.
- Public Health Direction, Institut Pasteur de Dakar, Dakar 220, Senegal.
- World Reference Center for Emerging Viruses and Arboviruses, Institute for Human Infections and Immunity and Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX 77555, USA.
*These authors contributed equally to this work.
- Corresponding author
Running Title: Chikungunya Virus Genomic characterization during an outbreak in Kedougou 2023.
Keywords: Chikungunya, Kedougou, Southeastern Senegal, Outbreak, genomic characterization.
Title: Genomic characterization of a reemerging Chikungunya outbreak in Kedougou, Southeastern Senegal, 2023.
Idrissa Dieng1*, Bacary Djilocalisse Sadio1*, Alioune Gaye2, Samba Niang Sagne3, Marie Henriette Dior Ndione1, Mouhamed Kane1, Mamadou Korka Diallo3, Bocar Sow3, Safietou Sankhe1, Amadou Diallo3, Madeleine Dieng1, Serge Freddy Moukaha Doukanda1, Maimouna Mbanne1, Seynabou Mbaye Ba Souna Diop1, Diamilatou Balde1, Mignane Ndiaye1, Khalidou Djibril Sow4, Maryam Diarra3, Abdoulaye Sam5, Ababacar Mbaye6, Boubacar Diallo7, Yoro Sall5, Ousmane Faye7, Boly Diop5, Abdourahmane Sow7, Amadou Alpha Sall1, Cheikh Loucoubar3, Ndongo Dia1, Oumar Faye1, Diawo Diallo2, Gamou Fall1, Scott C Weaver8, Mamadou Aliou Barry3, Mawlouth Diallo2, Moussa Moise Diagne1+
- Virology Department, Institut Pasteur de Dakar, Dakar 220, Senegal.
- Zoology Medical Department, Institut Pasteur de Dakar, Dakar 220, Senegal.
- Epidemiology, Clinical Research and Data Science Department, Institut Pasteur de Dakar, Dakar 220, Senegal.
- Health Emergency Operations Center, Ministry of Health, Dakar 220, Senegal.
- Prevention Department, Ministry of Health, Dakar 220, Senegal.
- Kedougou Medical Region, Ministry of Health, Kedougou 26005, Senegal.
- Public Health Direction, Institut Pasteur de Dakar, Dakar 220, Senegal.
- World Reference Center for Emerging Viruses and Arboviruses, Institute for Human Infections and Immunity and Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX 77555, USA.
*These authors contributed equally to this work.
Abstract
Chikungunya virus has caused millions of cases worldwide over the last twenty years, with recent outbreaks in Kedougou region in the southeastern Senegal, West Africa. Genomic characterization highlights that an ongoing epidemic in Kedougou in 2023 is not due to an introduction event but caused by the re-emergence of an endemic strain evolving linearly in a sylvatic context.
Background
Chikungunya virus (CHIKV) is a mosquito-borne virus that has caused significant epidemics
over the past 20 years with millions of cases reported worldwide. CHIKV is classified into West African (WA), East-Central-South-African (ECSA), Asian genotypes and Indian Ocean lineage (1). In Senegal, gallery forest mosquitoes maintain a sylvatic transmission cycle where sporadic cases or small outbreaks can occur among humans living in rural areas, while the virus is mainly transmitted by Aedes aegypti in urban settlements (2,3). Similarly to other countries in Africa and Asia, a cyclic CHIKV re-emergence or recurrence was observed in young, naive populations from rural areas after more or less a decade of silence. This interval is assumed to be the time required for turnover in susceptible vertebrate hosts, primarily non-human primates (4,5). Since its first isolation from a bat in 1962, sporadic outbreaks have been regularly reported (5), the most recent ones being in the Kedougou region, southeastern Senegal (2,6).
Kedougou is a major arbovirus hotspot as highlighted by an extensive surveillance through a nationwide syndromic network and a passive surveillance in public health structures coordinated by the ministry of health and the Institut Pasteur de Dakar (IPD) (7) as well as a long-term entomological surveillance (3). Moreover, the region has recently experienced an economic boom with the intensification of gold mining activity in rural area, causing migration of human populations from various horizons and subsequent environment changes with an increased risk of pathogens exposition.
Here we carry out the genomic characterization of the CHIKV strain of an ongoing epidemic in Kedougou in 2023.
The study
On early August 2023, a cluster of five Chikungunya virus-infected patients in the Kedougou region were identified by one-step RT-qPCR assay (2). Following the increasing number of cases, an investigation team of both MoH epidemiologists and IPD multidisciplinary group was mobilized to cover the three health districts of the region (Saraya, Salemata and Kedougou). At the time of writing, more than 200 confirmed cases were reported in the region and the outbreak is expanding to other regions (Communication from the IPD Public Health Direction).
Whole blood samples from suspected cases identified during the investigations were collected in the different healthcare sites and sent to the IPD station in Kedougou for CHIKV molecular diagnostic before transmission to the WHO collaborating Center for Arboviruses and Hemorrhagic Fever Viruses (CRORA) in IPD in Dakar for complementary laboratory analysis, as previously described (6). Meanwhile, mosquitoes collected from the entomological investigations in Kedougou were also tested onsite for CHIKV infection.
Nine human and three mosquito samples among the positive samples were randomly selected for genomic characterization. Samples were processed to obtain the whole genome by a target enrichment standard hybridization workflow using the Twist Biosciences Comprehensive Viral Research Panel (CVRP) as previously described (8). Briefly, extracted RNA samples were used as a template for a reverse transcription step with the SuperScript IV Reverse Transcriptase kit (Invitrogen, Thermo Fisher, USA) followed by a DNA fragmentation, telomere repair, dA-Tailing and a ligation with Universal Twist adapters before a final libraries amplification. A single pooled library was finally prepared from the indexed library-prepped samples before hybridization of the targets in solution, then binding of hybridized targets to desired streptavidin beads. Enriched sample libraries obtained as recommended by Twist Technical Support was loaded onto a Illumina iSeq 100 sequencing system as recommended by the manufacturer. Since no CHIKV sequence from the Kedougou 2015 outbreak was available, eight isolates obtained from the CRORA biobank were sequenced for further analysis.
Indeed, all generated sequences during this work (n = 20) were aligned with a representative dataset of available CHIKV sequences (Table S1) using MAFFT with default parameters and a maximum likelihood tree was subsequently performed using IQ-TREE with the best model determined by ModelFinder as previously described (8). In parallel a second 1,000 iterations based ML tree built with only WA genotype strains was used as input for root to tip analysis using Tempest (9). Amino acid (aa) changes spanning coding region of newly generated CHIKV sequences against human sequence obtained in 2005 in Kedougou (HM045817) were called using a python script according to a previously described protocol (10).
Phylogenetic tree showed that viral strain from Kedougou 2023 groups with previous CHIKV from the WA genotype identified during the more recent outbreaks in Kedougou in 2005 and 2015 (figure 1). Indeed, blast analysis showed that the circulating strains in 2015 and 2023 exhibit respectively mean nucleotide difference of 98.96% and 98.80% with HM045817 strain obtained from human in Kedougou in 2005 (Table S2).
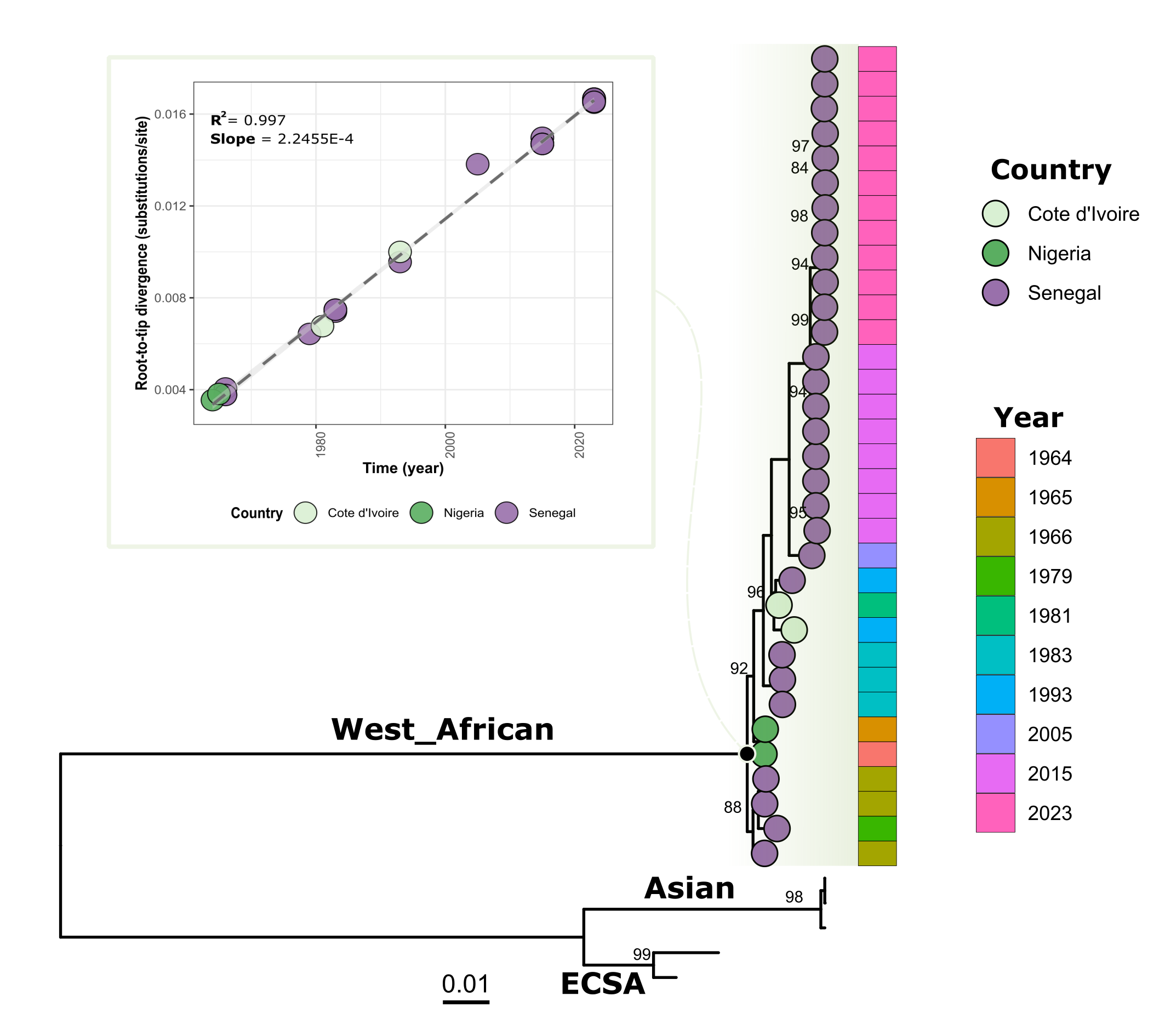
Figure 1: Phylogenetic analysis of newly generated CHIKV sequences obtain from ongoing outbreak in Senegal 2023 (n = 12) and those from 2015 outbreak in Kedougou area (n = 08).
Pane A) shows the correlation between sampling time and genetic divergence of WA genotype CHIKV sequences; Pane B) Shows the phylogenetic relationship of newly generated CHIKV sequences an those previously available. Tree was annotated with country and year of sampling of West African genotype viral sequences. Only bootstrap values above 70 were represented on corresponding branches.
The polyprotein molecular analysis demonstrated 28 aminoacids (aa) changes in both structural and nonstructural genes among either strains from CHIKV 2015 and CHIKV 2023 outbreaks in Kedougou (Table 1).
Table 1 : Non synonymous mutations observed in both structural and nonstructural genes in Chikungunya virus strains from 2015 and 2023 in Kedougou. CHIKV genome from 2005 in Kedougou (HM045817) was taken as reference.
Table 1.pdf (58.1 KB)
CHIKV 2015 and CHIKV 2023 polyproteins shared twelve aa substitutions compared to the older CHIKV 2005. Most of them are novel mutations identified in E1, E2 and nsP1-4, with E2-M92T, E2-S380T and nsP2-N580S in sites previously associated with type I Interferon modulation, antigenicity and host receptors binding (11, 12). CHIKV 2023 harbors eight unique mutations not seen in the CHIKV 2005 and 2015 strains, among which only two consistently found in nonstructural proteins in all the generated sequences. Indeed, nsP1-P57S was found in the N-terminus (NT) methyltransferase and guanylyltransferase domain involved in methylation and capping of the newly synthesized RNA (13), while nsP4-I603V was noted in the gene encoding the RNA-dependent RNA polymerase responsible for replicating viral RNA (14). The six other CHIKV 2023-specific aa mutations (C-I167V, E1-I280V, E2-H147Q, nsP1-S259T, nsP2-A674V and nsP4-A462T) occurred with lower frequency (8%-59%) as shown in table 1, and could then be either transient deleterious mutations which will be purged by purifying selection or potential new major variants whose prevalence is expected to increase as the virus expands.
Overall, the genomic analysis of our study reveals that the ongoing CHIKV epidemic in Kedougou in 2023 is not caused by the introduction of a new CHIKV strain but by the re-emergence of an endemic WA genotype strain having evolved linearly (R = 0.99) (Figure 1; Pane A) in a sylvatic context before a spillover event in the rural domain.
Indeed, our genomic characterization work demonstrates the endemicity of CHIKV in eastern Senegal and the constant threat it poses in terms of public health with cyclic resurgences. Indeed, the pathogen responsible for the 2023 epidemic arises from the regular molecular evolution of the strains from the 2005 and 2015 outbreaks. This supports previous work which highlights that the particular ecology of the Kedougou region, where human settlements overlap with the wild environment, allows the maintenance of the virus via potential non-human reservoirs and sylvatic vectors before spillover events (4,5). Given that the main vectors during this epidemic are sylvatic (data not shown), it is of interest to evaluate the impact of the different aa substitutions in the virus adaptation to the mosquito species in the region, similarly to what was done with the A226V amino acid adaptative mutation in the E1 envelope glycoprotein of the ECSA genotype to Ae. albopictus in 2005 (15).
If the contemporary strains presented common features with the causative agents of the previous outbreaks, identification of the phenotypic association with the CHIKV 2023-specific aa changes requires more genomic, epidemiological and experimental data.
References
- de Souza WM, de Lima STS, Simões Mello LM, Candido DS, Buss L, Whittaker C, et al. Spatiotemporal dynamics and recurrence of chikungunya virus in Brazil: an epidemiological study. Lanc Micr. 2023;4,5.
- Diallo D, Sall AA, Buenemann M, Chen R, Faye O, Diagne CT, et al. Landscape ecology of sylvatic chikungunya virus and mosquito vectors in southeastern Senegal. PLoS Negl Trop Dis. 2012;6,6.
- Diallo M, Thonnon J, Traore-Lamizana M, Fontenille D. Vectors of Chikungunya virus in Senegal: current data and transmission cycles. Am J Trop Med Hyg. 1999;60,2:281-286.
- Althouse BM, Guerbois M, Cummings DAT, Diop OM, Faye O, Faye A, et al. Role of monkeys in the sylvatic cycle of chikungunya virus in Senegal. Nat Commun. 2018;9,1,1046.
- Sow A, Nikolay B, Faye O, et al. Changes in the Transmission Dynamic of Chikungunya Virus in Southeastern Senegal. Viruses. 2020;12,2,196.
- Sow A, Faye O, Diallo M, Diallo D, Chen R, Faye O, et al. Chikungunya Outbreak in Kedougou, Southeastern Senegal in 2009-2010. Open Forum Infect Dis. 2017;2,5,1.
- Diagne MM, Ndione MHD, Gaye A, Barry MA, Diallo D, Diallo A, et al. Yellow Fever Outbreak in Eastern Senegal, 2020-2021. Viruses. 2021; 28,13,8:1475.
- Crispell G, Williams K, Zielinski E, Iwami A, Homas Z, Thomas K. Method comparison for Japanese encephalitis virus detection in samples collected from the Indo-Pacific region. Front Public Health. 2022; 24,10:1051754.
- Rambaut A, Lam TT, Max Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016;9,2,1:vew007.
- Dieng I, Balde D, Talla C, Camara D, Barry MA, Sagne SN, et al. Molecular Evolution of Dengue Virus 3 in Senegal between 2009 and 2022: Dispersal Patterns and Implications for Prevention and Therapeutic Countermeasures. Vaccines (Basel). 2023;28,11,10:1537.
- Sahu A, Das B, Das M, Patra A, Biswal S, Kar SK, Hazra RK. Genetic characterization of E2 region of Chikungunya virus circulating in Odisha, Eastern India from 2010 to 2011. Infect Genet Evol. 2013;18:113-24.
- Liu X, Mutso M, Cherkashchenko L, Zusinaite E, Herrero LJ, Doggett SL, et al. Identification of Natural Molecular Determinants of Ross River Virus Type I Interferon Modulation. J Virol. 2020;31,94,8:e01788-19.
- Rana J, Rajasekharan S, Gulati S, Dudha N, Gupta A, Chaudhary VK, Gupta S. Network mapping among the functional domains of Chikungunya virus nonstructural proteins. Proteins. 2014;82,10:2403-11.
- Chen MW, Tan YB, Zheng J, Zhao Y, Lim BT, Cornvik T, et al. Chikungunya virus nsP4 RNA-dependent RNA polymerase core domain displays detergent-sensitive primer extension and terminal adenylyltransferase activities. Antiviral Res. 2017;143:38-47.
- Tsetsarkin KA, Weaver SC. Sequential adaptive mutations enhance efficient vector switching by Chikungunya virus and its epidemic emergence. PLoS Pathog. 2011;7,12:e1002412.
Acknowledgments
We thank the healthcare workers from the Senegalese ministry of health for their dedication in the outbreak management. We are also grateful to the teams from the different research department in Institut Pasteur de Dakar.
This work was funded by the NIH West African Center for Emerging Infectious Diseases (grant number U01AI151801-01), the NIH PICREID (grant number U01AI151758) and the Africa CDC Pathogen Genomics Initiative funds (CARES grant 4306-22-EIPHLSS-GENOMICS).
Informed consent
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Senegalese national ethics committee for health research (protocol code SEN20/08 approved on 6 April 2020).
Disclaimers
The opinions expressed by authors contributing to this journal do not necessarily reflect the opinions of the Centers for Disease Control and Prevention or the institutions with which the authors are affiliated.