Cardozo, Fátima1*; Rojas, Alejandra1*; Marquez, Sully2; Carvajal; Mateo2; Morel, Roque1; Bernal, Cynthia1; Galeano, María Eugenia1; Valenzuela, Adriana1; Waggoner, Jesse J3; Cárdenas, Paul2; Martínez, Magaly1**†**
1Universidad Nacional de Asunción, Instituto de Investigaciones en Ciencias de la Salud, San Lorenzo, Paraguay
2Universidad San Francisco de Quito, Instituto de Microbiología, Quito, Ecuador
3Emory University, School of Medicine, Atlanta, GA, United States
*These authors contributed equally to this work.
† Corresponding author: [[email protected]]
Chikungunya virus (CHIKV) is an arbovirus of epidemic concern that causes arthralgia and, potentially, severe complications in humans (Schwartz et al., 2010). Autochthonous transmission of CHIKV in Paraguay was confirmed in 2015. Specific outbreaks were registered in Asunción and the Central department in 2015 and 2016, and then in 2018 in the Amambay department (border with Brazil). No deaths were attributable to CHIKV from 2015 to 2021 (DGVS, SE 13_2023).
In March of 2023, an epidemiologic alert was issued due to the increase in cases and deaths from chikungunya in the Region of the Americas, indicating that Paraguay had the highest incidence rate (PAHO alert March). Between October 2022 and mid-March 2023, more than 40.000 cases were reported, with cases in all the departments of the country (WHO, 2023).
In this study, we sequenced 30 serum samples collected in Asunción and the Central Department in January 2023 by Oxford nanopore technology (ONT). The genome amplification was attempted by multiplex PCR (Quick, 2017). We generated 30 CHIKV near-complete genomes (average coverage n: 94,06% - CDS: 98,87%). Genome Detective tool assigned consensus sequences as East/Central/South African (ECSA) lineage.
Maximum Likelihood phylogeny showed that the current Paraguayan strains formed a monophyletic cluster separate from Paraguayan ECSA strains detected in 2018, indicating the introduction of a new clade into Paraguay. Moreover, we performed a root-to-tip genetic distance analysis using TempEst version 1.5.3 (Rambaut et al., 2016) and rooted the trees using the R2 method. Maximum clade credibility (MCC) tree (Figure 1) was estimated by constructing a Bayesian time-calibrated tree by using BEAST version 1.10.4 under a Hasegawa-Kishino-Yano substitution model and an uncorrelated lognormal relaxed molecular clock (Drummond et al., 2006). We used a Bayesian SkyGrid tree prior (Gill et al., 2013) with 50 parameters and the time at the last transition point of one. We ran Markov chain Monte Carlo chains for 100 million steps and logged trees every 1,000 steps; we discarded the first 10% of the trees as burn-in using TreeAnnotator version 1.10.4 (TreeAnnotator | BEAST Documentation). Considering effective sample sizes of >200 in Tracer version 1.7.1 (Rambaut et al., 2018), adequate mixing and convergence of model parameters were assessed. Maximum clade credibility (MCC) trees for each run were summarized using TreeAnnotator version 1.10.4, summarizing node ages as median heights.
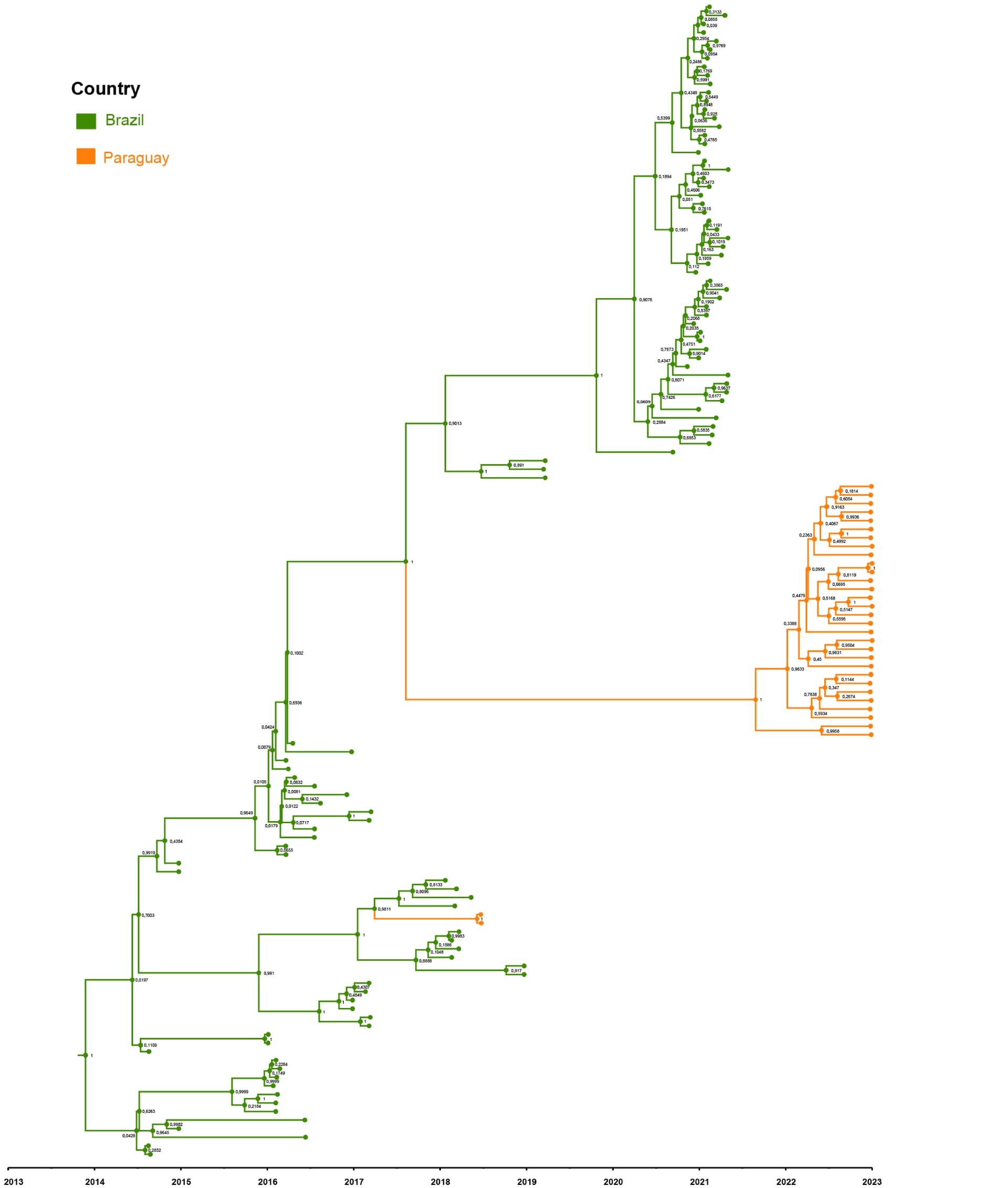
The MCC tree for CHIKV generated in BEAST software (http://beast.community) indicates two instances of viral variant exchange (viral exchanges) between Paraguay and Brazil dated back to 2017. In both instances, we found strong support for the persistence of one clade. In one instance, this viral exchange generated a new lineage which evolved until 2018 (clade I) and disappeared over time. Nevertheless, in the second instance (clade II), the introduction of those viruses into Paraguay is estimated to have occurred by 2021. The persistence of clade II lineage in Paraguay over time without the presence of other sequences from other countries suggests that Paraguay has sustained transmission that is continuing to evolve independent of external viral introductions.
Mutations at surface glycoproteins E1 and E2
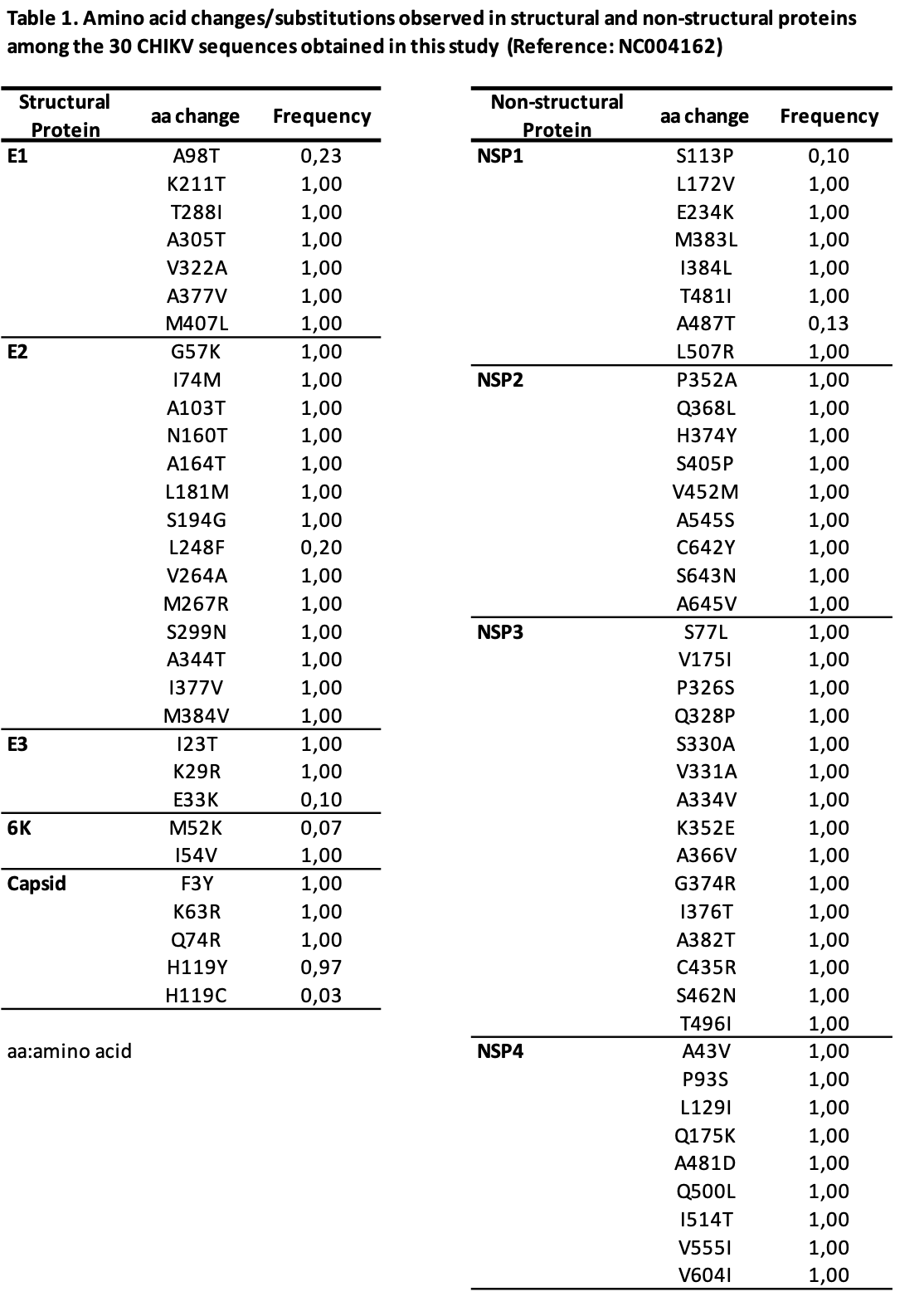
All structural proteins showed mutations relative to ECSA reference sequence NC004162 (Table 1). All the identified mutations have been detected in previous outbreaks in South America. Interestingly, substitutions related to vector fitness were identified:
E1:A98T: E1:98T occurred in 23,4% (7/30) sequences, while E1:98A was present in 76,6% (23/30). All Paraguayan ECSA sequences analyzed in this study harbored E1:226A. E1 and E2 are the two glycoproteins forming the chikungunya virion surface; they carry the main antigens and are responsible for attaching and entering the host cells. Through the evolution of CHIKV lineages, mutations associated with vector adaptability have been identified (Weaver, 2020). One of the best-characterized mutations in E1 that increased viral fitness for Aedes albopictus is E1:A226V, characteristic of the Indian Ocean Lineage (IOL), a descendant of the ECSA lineage (Tsetsarkin, 2007). The epistatic interaction with residue E1:98A strongly enhances the penetrance of this mutation, while E1:98T, mainly present in the Asian lineage, limits the adaptation to Ae. albopictus by constraining the acquisition of E1:226V (Tsetsarkin, 2011). Also, other mutations in E2 (e.g., L210Q, I211T and K252Q) increase the fitness of E1:A226V variants (Tsetsarkin, 2014). No E2 mutations associated with Ae. albopictus fitness were detected in this study.
E1:K211T and E2:V264A: Paraguayan ECSA strains sequenced for this study showed an E1:211T/E2:264A genotype, different from sequences that circulated during a minor outbreak in the country in 2018 that carried E1:211N/E2:264V (Gräf T, 2021).
In Paraguay, the species Aedes aegypti is predominant over Ae. Albopictus and is distributed across the country. The presence of Ae. Albopictus has been recorded in a few country departments but with low abundance (SENEPA-MSPBS, 2017). In this landscape, mutations that favor Ae. aegypti vectors will play an important role.
Since 2016, the ECSA lineage with E1:K211E/E2:V264A combination with an E1:226A background was predominant in some Asian outbreaks, identified mainly in Ae. Aegypti dominated regions, and this mutation combination has been described to increase titers in Ae*. aegypti* cells slightly (Agarwal A, 2016). An evolutionary analysis of ECSA lineages showed that E1:211 position is under positive selection (Khongwichit S, 2021). Another mutation, K211T was detected in Brazil during a large ECSA outbreak in Rio de Janeiro in 2015-2016 (Souza TMA, 2017), and predominated in ECSA sequences reported in successive outbreaks in Brazil (Souza TMA, 2019, Souza TMA, 2022). In 2022, Rangel et al. demonstrated that E1:K211T promotes CHIKV dissemination in Ae. aegypti mosquitoes and suggested it could influence cell attachment and pathogenesis (Rangel M, 2021). Regarding circulation of E2:V264A in South America, it was detected at low frequency in sequences from Brazil in 2019 (Fabri AA, 2020; Lazari CDS, 2023).
Mutations in non-structural proteins
All non-structural proteins presented non-synonymous substitutions (Table 1). Interestingly, we identified new mutations in NSP2, NSP3 and NSP4.
NSP2: Q368L and S405P at the helicase domain. Previous work proposed this region of CHIKV NSP2 to influence the interaction with RNA and hence viral replication (Law Y-S, 2021).
NSP3: A366V at the hypervariable domain (HVD). HVD on NSP3 of alphaviruses plays an essential role in interacting with host proteins necessary for viral replication. Specifically, it has been described to influence changes in NSP3 of chikungunya and other mosquito-borne alphaviruses on cellular stress granules response (Nowee G, 2021).
NSP4: L129 and Q175K at the RdRp domain . Studies on non-structural proteins of other arboviruses like Zika virus (ZIKV) demonstrated that NSP4 was a preferential site for positive selection. Changes are expected to modulate some aspects of viral fitness, either in mosquitoes or vertebrate hosts (Sironi M, 2016). Interestingly, Moreira et al., showed that the substitution NSP4:A481D in ECSA strains detected in Río de Janeiro in 2018 (and present in Paraguayan 2023 strains) presented signals of positive selection (Moreira F, 2023).
Further studies are needed to address whether the mutations observed in structural and non-structural proteins may have functional consequences for CHIKV vector/host adaptation, replication and/or infectivity, evasion from the immune system or pathogenesis, and its potential influence in outbreak magnitude.
This study reinforces that continued genomic surveillance strategies are needed to support the monitoring of CHIKV epidemics to better understand changes in the incidence and severity of the disease and to shed light on the highest outbreak caused by CHIKV in Paraguay.
Keywords: chikungunya virus, Paraguay, genomics
Financial support: This work was partially funded by the Solidarity Fund for Innovative Projects, Civil Society, Francophonie and Human Development (FSPI).
References
- · Schwartz, O., Albert, M. Biology and pathogenesis of chikungunya virus. Nat Rev Microbiol 8, 491–500 (2010)
- · Pan American Health Organization / World Health Organization. Epidemiological Alert: Increase in cases and deaths from chikungunya in the Region of the Americas. March 8th, 2023. Washington, D.C. PAHO/WHO. 2023 https://www.paho.org/es/documentos/alerta-epidemiologica-aumento-casos-defunciones-por-chikunguna-region-americas
- · DGVS-MSPyBS, Paraguay. Boletín Epidemiológico Semanal SE 13|2023
https://dgvs.mspbs.gov.py/boletin-epidemiologico-semanal/
- · World Health Organization. Geographical expansion of cases of dengue and chikungunya beyond the historical areas of transmission in the Region of the Americas [cited 2023 Apr 3]. https://www.who.int/emergencies/disease-outbreak-news/item/2023-DON448External
- · Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, Oliveira G, Robles-Sikisaka R, Rogers TF, Beutler NA, Burton DR, Lewis-Ximenez LL, de Jesus JG, Giovanetti M, Hill SC, Black A, Bedford T, Carroll MW, Nunes M, Alcantara LC Jr, Sabino EC, Baylis SA, Faria NR, Loose M, Simpson JT, Pybus OG, Andersen KG, Loman NJ. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc. 2017 Jun;12(6):1261-1276.
- · Weaver S, Chen R, Diallo M. Chikungunya Virus: Role of Vectors in Emergence from Enzootic Cycles. Annu. Rev. Entomol. 2020. 65:313–32
- · Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007 Dec;3(12):e201
- · Tsetsarkin KA, Weaver SC. Sequential adaptive mutations enhance efficient vector switching by Chikungunya virus and its epidemic emergence. PLoSPathog. 2011 Dec;7(12):e1002412.
- · Tsetsarkin, K., Chen, R., Yun, R. et al. Multi-peaked adaptive landscape for chikungunya virus evolution predicts continued fitness optimization in Aedes albopictus mosquitoes. Nat Commun 5, 4084 (2014).
- · Agarwal A, Sharma AK, Sukumaran D, Parida M, Dash PK. Two novel epistatic mutations (E1:K211E and E2:V264A) in structural proteins of Chikungunya virus enhance fitness in Aedes aegypti. Virology. 2016 Oct;497:59-68. doi: 10.1016/j.virol.2016.06.025. Epub 2016 Jul 15. PMID: 27423270.
- · Souza TMA, Azeredo EL, BadolatoCorrêa J, Damasco PV, Santos C, Petitinga Paiva F, Nunes PCG, Barbosa LS, Cipitelli MC, Chouin Carneiro T, Faria NRC, Nogueira RMR, de Bruycker Nogueira F, dos Santos FB. First Report of the East Central South African Genotype of Chikungunya Virus in Rio de Janeiro, Brazil. PLOS Currents Outbreaks. 2017 Feb 14. Edition 1
- · Souza TML, Vieira, Y.R., Delatorre, E. et al. Emergence of the East-Central-South-African genotype of Chikungunya virus in Brazil and the city of Rio de Janeiro may have occurred years before surveillance detection. SciRep 9, 2760 (2019).
- · Souza, U.J.B.d.; Santos, R.N.d.; Giovanetti, M.; Alcantara, L.C.J.; Galvão, J.D.; Cardoso, F.D.P.; Brito, F.C.S.; Franco, A.C.; Roehe, P.M.; Ribeiro, B.M.; Spilki, F.R.; Campos, F.S. Genomic Epidemiology Reveals the Circulation of the Chikungunya Virus East/Central/South African Lineage in Tocantins State, North Brazil. Viruses 2022, 14, 2311
- · Rangel MV, McAllister N, Dancel-Manning K, Noval MG, Silva LA, Stapleford KA. Emerging Chikungunya Virus Variants at the E1-E1 Interglycoprotein Spike Interface Impact Virus Attachment and Inflammation. J Virol. 2022 Feb 23;96(4):e0158621. doi: 10.1128/JVI.01586-21. Epub 2021 Dec 22.
- · Fabri, A.A.; Rodrigues, C.D.d.S.; Santos, C.C.d.; Chalhoub, F.L.L.; Sampaio, S.A.; Faria, N.R.d.C.; Torres, M.C.; Fonseca, V.; Brasil, P.; Calvet, G.; Alcantara, L.C.J.; Filippis, A.M.B.d.; Giovanetti, M.; de Bruycker-Nogueira, F. Co-Circulation of Two Independent Clades and Persistence of CHIKV-ECSA Genotype during Epidemic Waves in Rio de Janeiro, Southeast Brazil. Pathogens 2020, 9, 984.
- · Lázari CDS, Ramundo MS, Ten-Caten F, Bressan CS, de Filippis AMB, Manuli ER, de Moraes I, Pereira GM, Côrtes MF, Candido DDS, Gerber AL, Guimarães AP, Faria NR, Nakaya HI, Vasconcelos ATR, Brasil P, Paranhos-Baccalà G, Sabino EC. Clinical markers of post-Chikungunya chronic inflammatory joint disease: A Brazilian cohort. PLoSNegl Trop Dis. 2023 Jan 6;17(1):e0011037.
- · Gräf T, Vazquez C, Giovanetti M, de Bruycker-Nogueira F, Fonseca V, Claro I, et al. Epidemiologic History and Genetic Diversity Origins of Chikungunya and Dengue Viruses, Paraguay. Emerg Infect Dis. 2021;27(5):1393-1404.
- · Law Y-S, Wang S, Tan YB, Shih O, Utt A, Goh WY, Lian B-J, Chen MW, Jeng U-S, Merits A., Luo D. 2021. Interdomain flexibility of chikungunya virus nsP2 helicase-protease differentially influences viral RNA replication and infectivity. J Virol 95:e01470-20.
- · Nowee G, Bakker JW, Geertsema C, Ros VID, Göertz GP, Fros JJ, Pijlman GP. A Tale of 20 Alphaviruses; Inter-species Diversity and Conserved Interactions Between Viral Non-structural Protein 3 and Stress Granule Proteins. Front Cell Dev Biol. 2021 Feb 11;9:625711. doi: 10.3389/fcell.2021.625711.
- · Sironi M, Forni D, Clerici M, Cagliani R. Nonstructural Proteins Are Preferential Positive Selection Targets in Zika Virus and Related Flaviviruses. PLoSNegl Trop Dis. 2016 Sep 2;10(9):e0004978. doi: 10.1371/journal.pntd.0004978.
- · Moreira f, Menezes M, Salgado-Benvindo C, Whittaker C, et al. Epidemiological and genomic investigation of chikungunya virus in Rio de Janeiro state, Brazil, between 2015 and 2018. Epidemiological and genomic investigation of chikungunya virus in Rio de Janeiro state, Brazil, between 2015 and 2018. MedRxiv 2023. 2023.04.12.23288482;doi:https://doi.org/10.1101/2023.04.12.23288482
- · SENEPA - MSPyBS. PLAN DE MANEJO INTEGRADO DE VECTORES. 2017 https://senepa.gov.py/materiales/
Rambaut A, Lam TT, Max Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016;2:vew007. 10.1093/ve/vew007 - · Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:e88. 10.1371/journal.pbio.0040088
- · Gill MS, Lemey P, Faria NR, Rambaut A, Shapiro B, Suchard MA (2013) Mol Biol Evol 30, 713-724 [SkyGrid Coalescent].
- · Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol. 2018;67:901–4. 10.1093/sysbio/syy032