Philippe Selhorst1 & Antonio Mauro Rezende1, Tessa de Block1, Sandra Coppens1, Hilde Smet1, Joachim Mariën2, Anne Hauner1, Isabel Brosius1, Laurens Liesenborghs1, Emmanuel Bottieau1, Eric Florence1, Eugene Bangwen1, Kevin K Ariën1,2, Marjan Van Esbroeck1, Chris Kenyon1, Koen Vercauteren1
1 Institute of Tropical Medicine, Antwerp, Belgium
2 University of Antwerp, Antwerp, Belgium
[email protected]
Monkeypox virus (MPXV) is a zoonotic virus that circulates in a yet unidentified animal reservoir in Africa with humans and primates being considered incidental hosts. The virus belongs to the same genus as the variola virus, which causes smallpox, and presents with a similar presentation of a febrile prodrome and a characteristic rash. A case fatality rate of up to 10% in children has been reported, but is much lower in adults.
Although MPXV is usually constrained to small outbreaks in remote parts of Central and West African countries, with cases elsewhere mostly linked to Africa travel, several cases of monkeypox virus are recently and increasingly being detected around the world (United Kingdom, Portugal, Spain, Sweden, Italy, Canada, Belgium, Australia, France, Germany, and the USA). This raises concerns about increased human-to-human transmission and necessitates urgent investigation into the underlying causes.
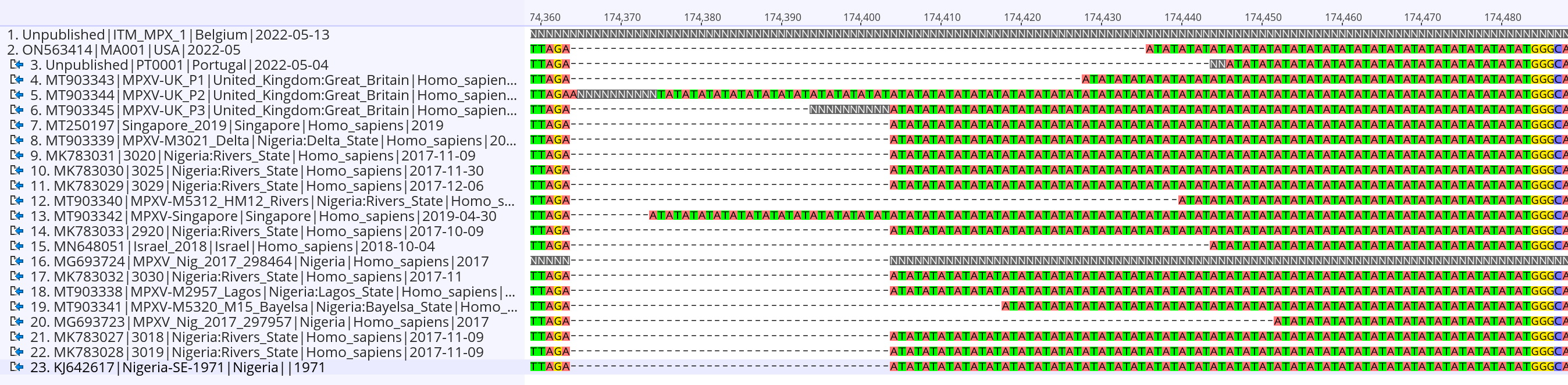
Here we report the nearly full genome of a 30-yr old male case in Belgium with a travel history to Lisbon, Portugal who presented to the ITM STI clinic on May 13th with perianal papules and 1cm painful inguinal adenopathy bilaterally. His partner since developed similar symptoms. Initial investigations of a sample taken from the lesions were negative for Herpes Simplex Virus and Treponema pallidum. Upon international alerts on MPX cases in Europe, additional analyses for MPXV returned positive (Ct of 16) with a MPXV qPCR using the generic G2RG_G primers (Li et al., 2010). Whole genome sequence data was obtained using an untargeted metagenomics approach, including a sequence-independent single primer amplification step, on Oxford Nanopore’s minION platform. We could reconstruct 98.9% of the genome with an average depth of 18.9x. Preliminary phylogenetic analysis clearly shows that the obtained genome belongs to the West African clade of MPXV and is most closely related to the recently uploaded genome from the outbreak in Portugal providing further evidence of substantial community spread in Europe.

The above phylogenetic analysis was performed by core alignment (using parsnp) of unambiguous regions from a rapid alignment of the newly sequenced genome with the publicly available genomes selected by Isidro et al (https://pando.tools/t/first-draft-genome-sequence-of-monkeypox-virus-associated-with-the-suspected-multi-country-outbreak-may-2022-confirmed-case-in-portugal/799). The Belgian and Portuguese sequence are depicted in red and clusters with sequences from a Nigerian 2018 outbreak which was exported to the UK, Israel, and Singapore.
Li, Y., Zhao, H., Wilkins, K., Hughes, C., & Damon, I. K. (2010). Real-time PCR assays for the specific detection of monkeypox virus West African and Congo Basin strain DNA. Journal of Virological Methods, 169(1), 223–227. https://doi.org/10.1016/j.jviromet.2010.07.012
Genome sequence updated on 2022-05-30:
ITM001_v2.zip (52.6 KB)