Keywords: Monkeypox virus, Mpox, Outbreak, Human-to-Human, Genomic, APOBEC3, Kinshasa, DRC.
The ongoing mpox outbreaks in the Democratic Republic of Congo (DRC) has raised significant public health concerns, particularly due to the emergence of the newly identified viral subclade Ib. Recently, we reported human-to-human transmission through sexual contact involving this subclade (1). This transmission route may be an important factor contributing to the virus’s spread from its initial hotspot in the Kamituga mining area to the densely populated cities of Bukavu and Goma, as well as nearby refugee camps in Eastern DRC (2). These developments, along with cross-border exportations within the region and outside of Africa (3), has led the WHO to declare mpox a Public Health Emergency of International Concern (PHEIC) for the second time on August 14th, 2024 (4, 5). Beyond the notable outbreak history of subclade Ib, we demonstrated that the DRC is experiencing multiple parallel outbreaks of various subclade Ia variant lineages, referred to as ‘groups’ (Group II, III, IV and V) (6). While these outbreaks have predominantly been driven by zoonotic transmission, instances of sexual transmission have also been observed (7). Although the areas most heavily impacted by the outbreak have been outside of Kinshasa, cases have also been reported within the capital (6, 8). Local secondary transmission observed during contact tracing in Kinshasa had remained limited, until now.
To better understand the ongoing situation, we sequenced samples from mpox-confirmed cases in Kinshasa, generating 115 near-complete Monkeypox virus (MPXV) genomes, and performed a phylogenomic analysis (Figure 1). This reveals an escalation of the Kinshasa mpox outbreak, involving the establishment of both subclade Ia (Group II) and subclade Ib MPXV. Subclade Ia introductions (n=11 samples belonging to 5 clusters within Group II) into Kinshasa were observed in 2023 and 2024 without established local transmission chains.
Due to the high fidelity and error correction of the MPXV DNA polymerase, mutations during genome replication occur at a much lower rate than those induced by APOBEC3 (~1 nucleotide change every 3 years vs ~6 per year respectively). Thus, over a short timescale during human-to-human transmission, we expect APOBEC3-type mutations to predominate and occur at a sufficient rate to reconstruct the patterns of transmission and spread of the virus. In this analysis, we estimate that 63% of mutations in subclade Ia (Figure 2) and 66% of mutations in subclade Ib (Figure 3) are consistent with APOBEC3-driven changes, a hallmark of human-to-human transmission (9). Although sequencing can introduce errors that manifest as artifactual mutations, these are unlikely to resemble the specific characteristics of those induced by APOBEC3. A small fraction of polymerase-induced mutations may, by chance, appear similar to APOBEC3-induced mutations (~ 8% is estimated for Clade I (9)), but their absolute rate is negligible over short timelines. Given the predominance of APOBEC3-type mutations in these outbreaks, specifically in the Clade Ia outbreak in Kinshasa and the entire Clade Ib outbreak first observed in Sud-Kivu, we can be confident that they both resulted from sustained human-to-human transmission (following a single zoonotic introduction). This represents a shift in the epidemiology of subclade Ia group outbreaks, which had been suggested to be driven by multiple independent zoonotic introductions, until now. This outbreak should, therefore, be closely monitored, as it represents a significant threat for regional and international dissemination as illustrated with new sequences from Kwilu, Kwango, and Kongo-Central (subclade Ia), as well as Kasaï, Tanganyika and Tshopo (subclade Ib), linked to Kinshasa genomes. The potential link with sexual networks should be further examined.
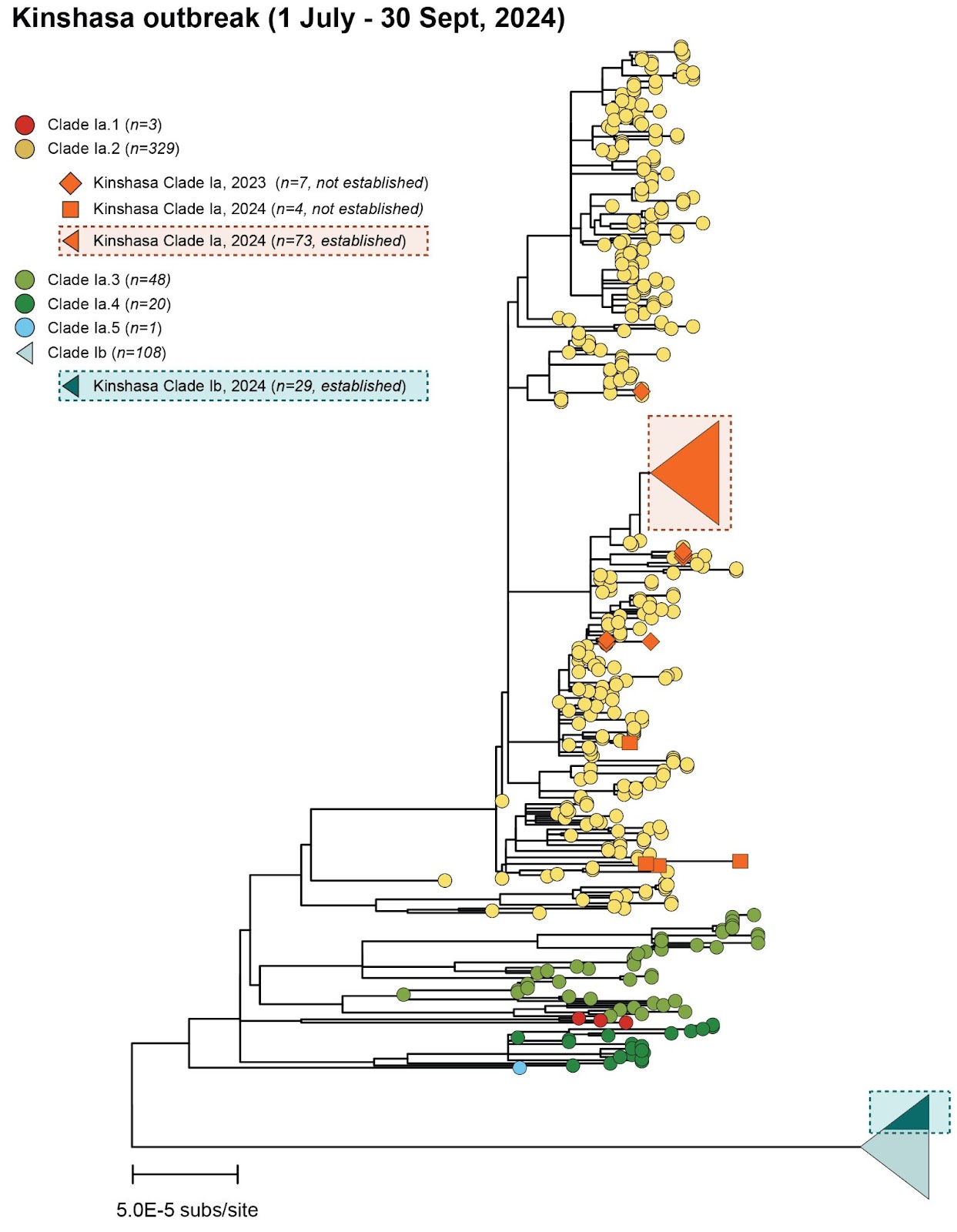
Figure 1 | Maximum likelihood phylogeny: Clade Ia as well as the recently discovered Clade Ib MPXV have reached Kinshasa, the capital of the DRC, to cause two co-emerging local outbreaks. The Maximum likelihood phylogeny shows that viral genomes isolated from mpox patients residing in Kinshasa, in 2023-2024 belong to subclade Ia (Orange, total n=93; established n=73; not established n=11) and Ib (Green, established n=29). Non-established Kinshasa subclade Ia observations are indicated with orange diamonds (2023) and squares (2024). Clade Ia ‘groups’ are indicated as: I (red), II (yellow), III (light green), IV (dark green), V (blue). The (established) Kinshasa Ia and Ib outbreaks are marked with rectangles with dashed borders.
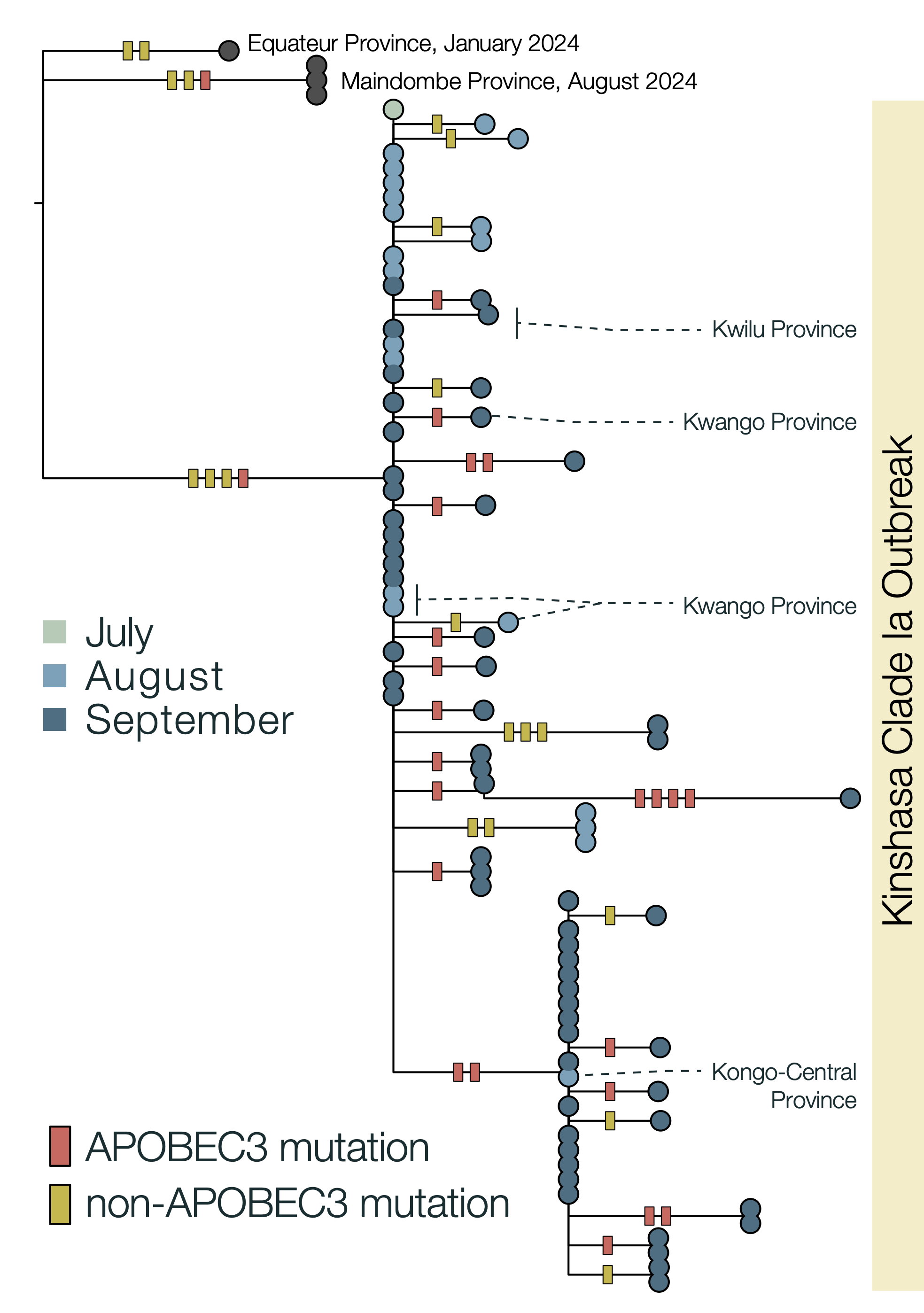
Figure 2 | Maximum likelihood tree and APOBEC3 reconstruction of a Kinshasa 2024 outbreak of MPXV Clade Ia. 63% (22/35) of the mutations in the ongoing 2024 Kinshasa subclade Ia outbreak, indicative of APOBEC3-induced changes in sustained human-to-human transmission. The Clade Ia Kinshasa outbreak genomes are shown annotated by month of sample collection in 2024. APOBEC3 driven changes are indicated by red bars on the branches, others are indicated as yellow bars. Outgroup genomes are indicated by black dots.
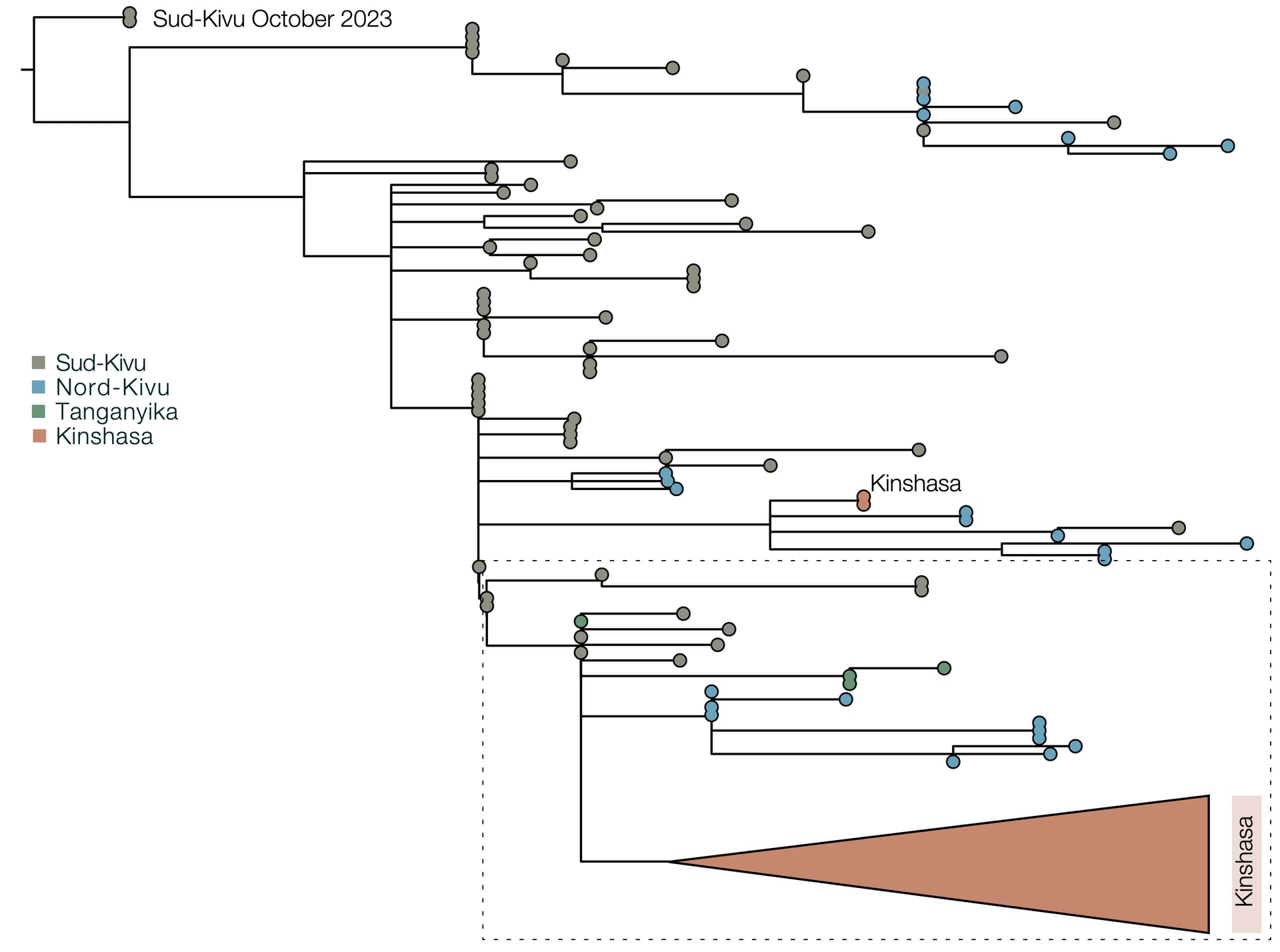
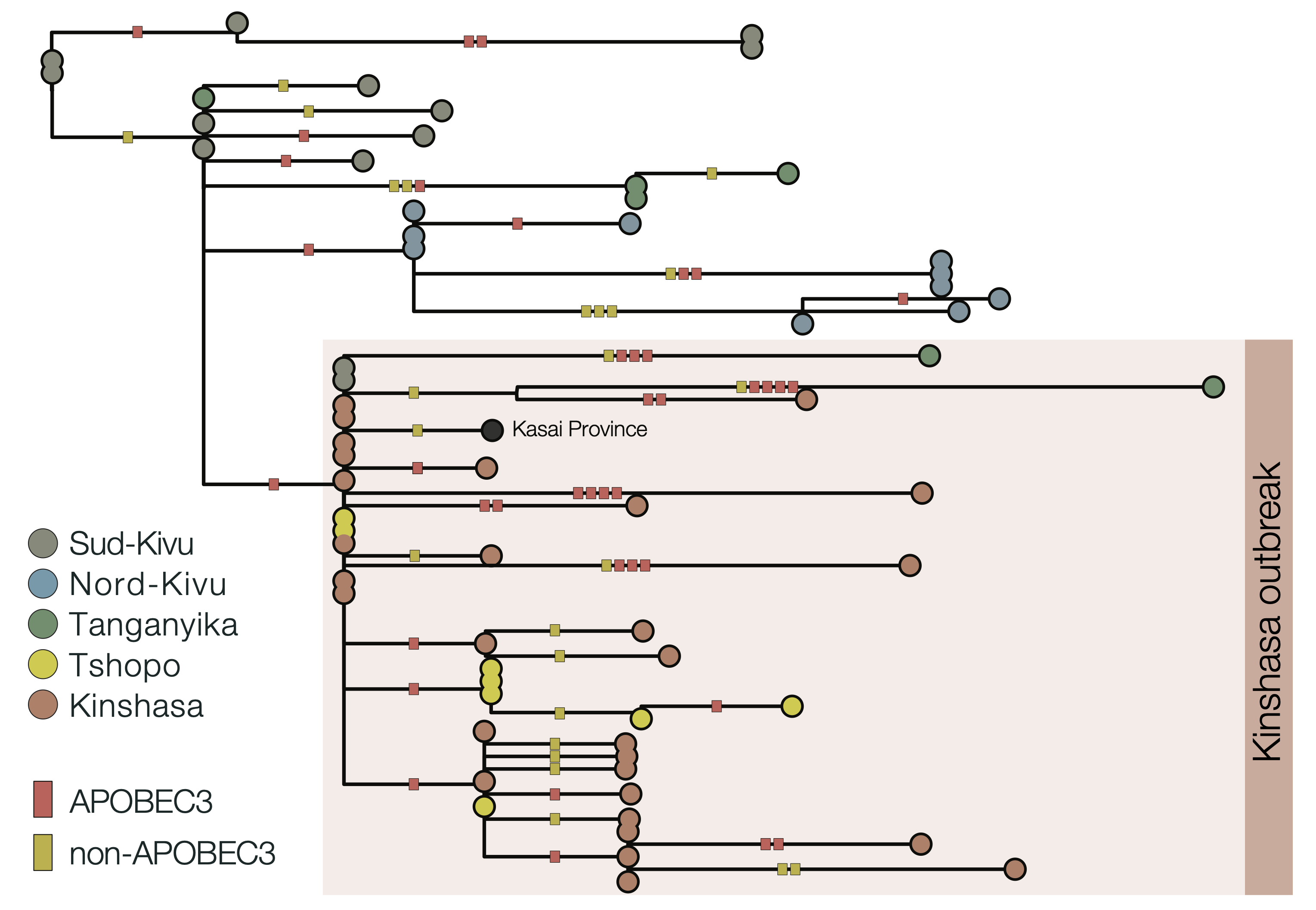
Figure 3 | Maximum likelihood phylogeny of country-wide subclade Ib outbreak (top panel) and APOBEC3 analysis of subclade Ib, Kinshasa 2024 outbreak (bottom panel): 66% (29/44) of the mutations in the ongoing 2024 Kinshasa subclade Ib outbreak (within the box) are consistent with APOBEC3-driven changes. This is consistent with the 60% (88/146) mutation fraction observed for the Ib outbreak overall (not shown). 2024 Kinshasa subclade Ib genomes are shown annotated by province (Sud-Kivu, Nord-Kivu, Tanganyika, Tshopo, and Kinshasa, with one additional genome from Kasaï Province). APOBEC3 driven changes are indicated by red bars on the branches, others are indicated as yellow bars. We annotated using the province instead of month of sample collection to highlight the geographic interconnectivity of the Kinshasa subclade Ib outbreak, especially with Tshopo province.
Methods
Sampling and Data Collection
Kinshasa, which is both a city and a province, is divided into 35 health zones in terms of healthcare services. A multidisciplinary team comprising representatives from the Ministry of Public Health, the Institut National de Santé Publique (INSP) and the Institut National de Recherche Biomédicale (INRB) has investigated the mpox outbreak in Kinshasa. Local surveillance teams collected data using the national case investigation form, as well as samples from mpox suspected cases which were sent to the Institut National de Recherche Biomédicale (INRB), Kinshasa for molecular confirmation and viral whole genome sequencing.
Mpox diagnosis and Whole genome sequencing
Viral DNA was extracted (QIAamp DNA Mini Kit, Qiagen, Hilden, Germany), before real-time PCR (Orthopoxvirus- and MPXV-generic primers/probes as described (12, 13)). Confirmed samples were subjected to sequencing using either hybridization capture probe-based (Comprehensive Viral Research Panel, Twist Biosciences) or amplicon-based (14) enrichment before loading onto either the MiSeq or GridION sequencer. FASTQ files were processed using GeVarLi (TRANSVIHMI / fernandez-nicolas / GeVarLi · GitLab), CZ ID (https://czid.org/), or an in-house pipeline (Metatropics) pipeline for Reference-based (GenBank ID: NC_003310) consensus genome generation.
Phylogenomic analysis of monkeypox virus
Clade I MPXV reference genomes were obtained from NCBI GenBank, and Lusamaki et al. (6) (accessed on 05/10/2024). We also included newly generated sequences from the ongoing national genomics surveillance (July-September: as available in github). Duplicate sequences were removed, and the dataset was filtered to include MPXV genomes meeting specific quality criteria: over 80% horizontal coverage (fewer than 20% ambiguous bases), exclusion of sequences flagged by IQ-TREE (14), and complete metadata for collection date and location. This dataset was then combined with consensus genomes from Clade I mpox cases in Kinshasa, DRC, using the same quality thresholds. The Clade IIa MPXV sequence (accession number: AY603973) was included as an outgroup. The entire MPXV genome dataset was aligned to the Clade I reference (accession: NC_003310) using Squirrel (GitHub - aineniamh/squirrel), with low-complexity and repetitive regions masked from the alignment. A maximum-likelihood phylogenetic tree was inferred using IQ-TREE2 v2.1.4 (15), with the ‘K3Pu+F+I+R5’ substitution model selected as the best fit. Branch support was determined through ultrafast bootstrap approximation with 10,000 replicates (Figure 1).
We then focused on the established Kinshasa Clade I MPXV outbreak genomes and reconstructed individual mutations onto each branch of the maximum likelihood tree, using ancestral state reconstruction in IQ-TREE2 (15). Squirrel then assigns these mutations into two types: non-APOBEC3-type mutations, caused by errors due to the polymerase at replication, and APOBEC3-type mutations, identified based on their dinucleotide context and specific transitions (9) (Figure 2 and 3).
Ethics considerations
Samples were collected as part of a routine country-wide mpox surveillance program and were therefore exempt from informed consent procedures. Permission to use anonymized data from the Mpox national surveillance activities for this report was granted by the Ethics Committee of the Kinshasa School of Public Health (ESP-UNIKIN, Number ESP/CE/05/2023).
Statement on continuing work and analyses prior to publication
Results of this study are based on ongoing work and should be considered preliminary. Our analyses are still in progress, and a publication detailing these findings, along with MPXV genomes, is currently in preparation. For further details or information, please contact Placide Mbala ([email protected]) and/or Tony Wawina-Bokalanga ([email protected]).
Partners and Authors’ affiliations
1. Institut National de Recherche Biomédicale (INRB), Kinshasa, Democratic Republic of the Congo: Tony Wawina-Bokalanga, Elisabeth Pukuta-Simbu, Emmanuel Hasivirwe Vakaniaki, Rilia Ola-Mpumbe, Prince Akil-Bandali, Papy Kwete-Mbokama, Adrienne Amuri-Aziza, Princesse Paku-Tshambu, Nelson Mapenzi, Emmanuel Lokilo, Chloé Muswamba, Leader Ontshick, Jean-Claude Makangara-Cigolo, Gradi Luakanda, Eddy Kinganda-Lusamaki, Steve Ahuka-Mundeke, Jean-Jacques Muyembe-Tamfum, Placide Mbala-Kingebeni
2. Service de Microbiologie, Département de Biologie Médicale, Cliniques Universitaires de Kinshasa, Université de Kinshasa: Tony Wawina-Bokalanga, Eddy Kinganda-Lusamaki, Jean-Claude Makangara-Cigolo, Steve Ahuka-Mundeke, Jean-Jacques Muyembe, Placide Mbala-Kingebeni
3. Institut National de Santé Publique (INSP), Kinshasa, Democratic Republic of the Congo: Cris Kacita, Christian Ngandu, Dieudonné Mwamba
4. Department of Clinical Sciences, Institute of Tropical Medicine, Antwerp, Belgium: Tony Wawina-Bokalanga, Daan Jansen, Pedro HLF Dantas, Tessa De Block, Laurens Liesenborghs, Koen Vercauteren
5. Institute of Ecology and Evolution, University of Edinburgh, Edinburgh, UK: Áine O’Toole, Andrew Rambaut
6. Department of Epidemiology, Jonathan and Karin Fielding School of Public Health, University of California, Los Angeles, CA, USA: Megan Halbrook, Sydney Merritt, Nicole A. Hoff, Anne W. Rimoin
7. TransVIHMI, Université de Montpellier, INSERM, IRD, Montpellier, France: Eddy Kinganda-Lusamaki, Ahidjo Ayouba, Eric Delaporte, Martine Peeters
8. Instituto René Rachou, Fiocruz Minas, Belo Horizonte, Brazil : Antonio Mauro Rezende
9. Institute of Social and Preventive Medicine, University of Bern, Bern, Switzerland: Nicola Low
10. Department of Medical Microbiology & Infectious Diseases, University of Manitoba, Winnipeg, Manitoba, Canada; Department of Internal Medicine, University of Manitoba, Winnipeg, Manitoba, Canada: Jason Kindrachuk
11. University of Birmingham, Birmingham, UK: Nick Loman, Sam Wilkinson, Josh Quick
12. Africa Centers for Disease Control and Prevention (Africa CDC), Addis Ababa, Ethiopia: Sofonias Tessema, Nicaise Ndembi
13. World Health Organization, Geneva, Switzerland: Lorenzo Subissi
References
-
Vakaniaki EH, Kacita C, Kinganda-Lusamaki E, O’Toole A, Wawina-Bokalanga T, Mukadi-Bamuleka D, et al. Sustained human outbreak of a new MPXV clade I lineage in eastern Democratic Republic of the Congo. Nat Med. 2024 Jun 13.
-
Mukadi-Bamuleka D, Kinganda-Lusamaki E, Mulopo-Mukanya N, Amuri-Aziza A, O’Toole A, Modadra-Madakpa B, et al. First imported Cases of MPXV Clade Ib in Goma, Democratic Republic of the Congo: Implications for Global Surveillance and Transmission Dynamics. medRxiv. 2024 Sep 16.
-
ECDC. Epidemiological update: Mpox due to monkeypox virus clade I. (2024). 2024.
-
Adepoju P. Mpox declared a public health emergency. Lancet. 2024 Aug 24;404(10454):e1-e2.
-
WHO. WHO Director-General declares mpox outbreak a public health emergency of international concern 14-08-2024. 2024.
-
Kinganda-Lusamaki E, Amuri-Aziza A, Fernandez N, Makangara-Cigolo J-C, Pratt C, Hasivirwe Vakaniaki E, et al. Clade I Mpox virus genomic diversity in the Democratic Republic of the Congo, 2018 - 2024: Predominance of Zoonotic Transmission. medRxiv. 2024:2024.08.13.24311951.
-
Kibungu EM, Vakaniaki EH, Kinganda-Lusamaki E, Kalonji-Mukendi T, Pukuta E, Hoff NA, et al. Clade I-Associated Mpox Cases Associated with Sexual Contact, the Democratic Republic of the Congo. Emerg Infect Dis. 2024 Jan;30(1):172-6.
-
Wawina-Bokalanga T, Akil-Bandali P, Kinganda-Lusamaki E, Lokilo E, Jansen D, Amuri-Aziza A, et al. Co-circulation of monkeypox virus subclades Ia and Ib in Kinshasa Province, Democratic Republic of the Congo, July to August 2024. Euro Surveill. 2024 Sep;29(38).
-
O’Toole A, Neher RA, Ndodo N, Borges V, Gannon B, Gomes JP, et al. APOBEC3 deaminase editing in mpox virus as evidence for sustained human transmission since at least 2016. Science. 2023 Nov 3;382(6670):595-600.
-
Jezek Z, Grab B, Szczeniowski M, Paluku KM, Mutombo M. Clinico-epidemiological features of monkeypox patients with an animal or human source of infection. Bull World Health Organ. 1988;66(4):459-64.
-
Van Dijck C, Hoff NA, Mbala-Kingebeni P, Low N, Cevik M, Rimoin AW, et al. Emergence of mpox in the post-smallpox era-a narrative review on mpox epidemiology. Clin Microbiol Infect. 2023 Dec;29(12):1487-92.
-
Li Y, Zhao H, Wilkins K, Hughes C, Damon IK. Real-time PCR assays for the specific detection of monkeypox virus West African and Congo Basin strain DNA. J Virol Methods. 2010 Oct;169(1):223-7.
-
Schroeder K, Nitsche A. Multicolour, multiplex real-time PCR assay for the detection of human-pathogenic poxviruses. Mol Cell Probes. 2010 Apr;24(2):110-3.
-
Chen NFG, Chaguza C, Gagne L, Doucette M, Smole S, Buzby E, et al. Development of an amplicon-based sequencing approach in response to the global emergence of mpox. PLoS Biol. 2023 Jun;21(6):e3002151.
-
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol. 2020;37(5):1530-4.