A novel, room temperature-stable, multiplexed RT-qPCR assay to distinguish lineage B.1.1.7 from the remaining SARS-CoV-2 lineages
Viera Kovacova1,†, Kristína Boršová2,3†, Evan D Paul1,†, Monika Radvanszka1, Roman Hajdu1, Viktória Čabanová2, Monika Sláviková2, Martina Ličková2, Ľubomíra Lukáčiková2, Andrej Belák4,5, Lucia Roussier5, Michaela Kostičová5,6, Anna Líšková7, Lucia Maďarová8, Mária Štefkovičová9,10, Lenka Reizigová9,11, Elena Nováková12, Peter Sabaka13, Alena Koščálová13,14, Broňa Brejová15, Tomáš Vinař16, Jozef Nosek17, Pavol Čekan1,* , Boris Klempa2,*
1 MultiplexDX, Bratislava, Slovakia
2 Biomedical Research Center, Institute of Virology, Slovak Academy of Sciences, Bratislava, Slovakia
3 Department of Microbiology and Virology, Faculty of Natural Sciences, Comenius University in Bratislava, Bratislava, Slovakia
4 Institute of Ethnology and Social Anthropology, Slovak Academy of Sciences, Bratislava, Slovakia
5 Intervention team, Ministry of Healh, Slovakia
6 Institute of Social Medicine and Medical Ethics, Faculty of Medicine, Comenius University in Bratislava, Bratislava, Slovakia
7 Nitra Faculty Hospital, Department of Clinical Microbiology, Nitra, Slovakia
8 Regional Authority of Public Health, Banska Bystrica, Slovakia
9 Regional Authority of Public Health, Trenčín, Slovakia
10 Faculty of Healthcare, Alexander Dubček University of Trenčín, Slovakia
11 Department of Laboratory Medicine, Faculty of Healthcare and Social Work, Trnava University, Trnava, Slovakia
12 Department of Microbiology and Immunology, Comenius University in Bratislava, Jessenius Faculty of Medicine, Martin, Slovakia
13 Department of Infectology and Geographical Medicine, Faculty of Medicine, Comenius University in Bratislava, Bratislava, Slovakia
14 Department of Infectious Diseases, Slovak Medical University, Bratislava, Slovakia.
15 Department of Computer Science, Faculty of Mathematics, Physics and Informatics, Comenius University in Bratislava, Bratislava, Slovakia
16 Department of Applied Informatics, Faculty of Mathematics, Physics and Informatics, Comenius University in Bratislava, Bratislava, Slovakia
17 Department of Biochemistry, Faculty of Natural Sciences, Comenius University in Bratislava, Bratislava, Slovakia
† These authors contributed equally
* Correspondence to: [email protected]
* Correspondence to: [email protected]
Key words: B.1.1.7 lineage, 20I/501Y.V1, Variant of Concern (VOC) 202012/01, SARS-CoV-2, COVID-19, RT-qPCR, spike gene, spike protein, coronavirus
Introduction
The recent emergence of a novel variant of SARS-CoV-2 called lineage B.1.1.7 has sparked global alarm due to evidence of increased transmissibility, mortality, and uncertainty about vaccine efficacy, thus accelerating worldwide efforts to detect and track the variant. Current approaches to detect the B.1.1.7 variant include sequencing and molecular tests that contain a target assay that fails or results in reduced sensitivity towards the B.1.1.7 variant. Since many countries lack a robust genomic surveillance program and the failed target assays detect multiple unrelated variants that contain similar mutations as B.1.1.7, there is an urgent need to develop molecular tests that can accurately and rapidly identify the B.1.1.7 variant. We have developed a room temperature-stable, multiplexed RT-qPCR test that readily differentiates the B.1.1.7 variant from the most common SARS-CoV-2 variants. The test consists of two assays that target either the common SARS-CoV-2 spike gene or the two deletions in the spike gene (ΔH69/ΔV70 and ΔY144) that are present in the B.1.1.7 variant. Moreover, a simple relative comparison of the Ct values of the two assays permits not only identification of the B.1.1.7 variant but also its differentiation from other variants that harbor only the ΔH69/ΔV70 deletion. Each assay is multiplexed with a human RNase P internal control to assess RNA extraction and assay performance. This test can easily be implemented in diagnostic labs to rapidly scale B.1.1.7 surveillance efforts and is particularly useful in countries with high prevalence of variants possessing only the ΔH69/ΔV70 deletion because current strategies using target failure assays incorrectly identify these as putative B.1.1.7 variants.
Results
Analysis of sequences
Our analysis of 1,136 spike gene sequences (spanning 1 - 21 December 2020) revealed 228 sequences (20%) that contained both the ΔH69/ΔV70 and ΔY144 deletions (for country of origin, see Table 1). The shorter deletion (ΔY144) always co-occurred with the longer deletion (ΔH69/ΔV70), whereas the (ΔH69/ΔV70) deletion occurs independently in 17 sequences (1.5%). Pearson’s correlation coefficient of the deletions is 0.953.
Analysis of the prevalence of both ΔH69/ΔV70 and ΔY144 deletions in lineages other than B.1.1.7 revealed a total of 29,872 sequences that possess both deletions, while 368,474 sequences do not have them. Based on the metadata file, we identified SARS-CoV-2 lineages across all called sequences with both deletions. Only five sequences (0.0167%) out of 29,872 records are not labelled as B.1.1.7. In other words, 99.98% of sequences containing both deletions belong to lineage B.1.1.7, highlighting the notion that these two deletions are highly specific for the B.1.1.7 variant and make ideal targets for primer/probe design.
Optimization of a S gene primer/probe set targeting both common SARS-CoV-2 and ΔH69/ΔV70 deletion variants
We began by designing a general S gene primer/probe assay (SARS-CoV-2 S gene) that could be used for screening purposes and would detect the most common strains of SARS-CoV-2 as well as variants containing the ΔH69/ΔV70, including lineage B.1.1.7. We designed a series of primers flanking the ΔH69/ΔV70 deletion and tested their performance during RT-qPCR using the wild type and ΔH69/ΔV70 templates (for primer/probes sequences, see Table 2). Although all primers/probe combinations amplified both wild type (Figure 1A) and ΔH69/ΔV70 (Figure 1B) templates, we selected the F1-P3-R1 set for further analysis since it resulted in the lowest overall Ct values and sufficient fluorescence intensity.
Dual probes enhance fluorescence intensity and sensitivity
Given that prior reports suggest that including a second TaqMan hydrolysis probe can be beneficial for specificity and sensitivity (Nagy et al., 2017; Yip et al., 2005), we designed an additional hydrolysis probe (P4), identically labelled with the same reporter and quencher dyes, that would hybridize in tandem (i.e., on the same strand) with the first hydrolysis probe (P3). We compared single versus dual hydrolysis probe assays using three 10-fold dilutions of template RNA and observed that dual probes resulted in greater fluorescent intensity at all dilutions (Figure 1C) as well as reduced Ct values at the first two dilutions (Figure 1D). Moreover, the efficiency of the RT-qPCR reaction was similar between single and dual probe assays. Therefore, we decided to incorporate dual probes into our SARS-CoV-2 S gene primer/probe set (F1-P3-P4-R1; Table 2)
Optimization of a S gene primer/probe set capable of differentiating between lineage B.1.1.7 and other SARS-CoV-2 variants
Lineage B.1.1.7 contains two deletions in the spike gene, including a six base pair deletion (bp: 21765-21770; ΔH69/ΔV70) and three base pair deletion (bp: 21991-21993; ΔY144). Since the co-occurance of these deletions is highly specific to B.1.1.7 (99.98%), we selected these deletions as candidate regions to target for a primer/probe set to differentiate B.1.1.7 from all other SARS-CoV-2 variants (see Table 3 for primer and probe sequences). Our first approach consisted of designing several probes targeting the first deletion and determining specificity by conducting RT-qPCR using the forward and reverse primers from our SARS-CoV-2 S gene set (F1 and R1) to amplify both wild type and ΔH69/ΔV70 templates; however, none of the probes were capable of discriminating between wild type and ΔH69/ΔV70 templates, even though all probes were less efficient at detecting wild type template (Figure 2A). We then attempted to differentiate wild type template from ΔH69/ΔV70 template by designing several forward primers targeting the span of the six base pair deletion. We tested these forward primers using the same probe and reverse primer from our SARS-CoV-2 S gene set (P3 and R1) and found two forward primers (F2 and F3) that specifically amplified only ΔH69/ΔV70 template (Figure 2B). We selected F3 instead of F2 for further assay development based on its sensitivity to ΔH69/ΔV70 templates, fluorescence intensity, and profile of the amplification curve.
Since other SARS-CoV-2 variants share the ΔH69/ΔV70 deletion (e.g., B.1.1.298, B.1.160, B.1.177, B.1.258, B.1.375), we next sought to design a series of reverse primers to target the second, three base pair deletion (bp: 21991-21993; ΔY144). This approach would facilitate differentiating B.1.1.7 variants that contain both the ΔH69/ΔV70 and ΔY144 deletions from SARS-CoV-2 variants that contain only the ΔH69/ΔV70 deletion. To increase the specificity of the assay, we also utilized allele-specific PCR approaches. We introduced selective mismatch bases in the 3’-end of the primer to destabilize base complementary to sequences that lack the ΔY144 deletion, while retaining complementarity in sequences containing the ΔY144 deletion such as B.1.1.7 variants. We designed a series of reverse primers spanning the three base pair deletion (bp: 21991-21993; ΔY144) with and without LNA-modified oligos to modify stability and tested the ability of these primer/probe sets to amplify three templates: wild type, ΔH69/ΔV70, and B.1.1.7. While our 3’-LNA-modified primers (R1-R6) were unable to differentiate between B.1.1.7 and ΔH69/ΔV70, several unmodified primers displayed reduced sensitivity towards ΔH69/ΔV70 with a Ct value difference greater than nine when compared with Ct values of B.1.1.7, demonstrating their capability to differentiate the two variants (Figure 2C). These primers also showed minimal reductions in sensitivity when compared to Ct values of B.1.1.7 template amplified using SARS-CoV-2 S gene set (Figure 2D). This data strongly suggests that the change in Ct value (ΔCt), that is to say, the relative difference in Ct values between the B.1.1.7 and SARS-CoV-2 S gene assays, could be used to confirm the presence of B.1.1.7 when the ΔCt value is minimal (e.g., ΔCt ≤ 5). Alternatively, a larger change in Ct values (e.g., ΔCt ≥ 8) could indicate the presence of a variant that contains the ΔH69/ΔV70 deletion, but not the ΔY144 deletion.
We also attempted another approach by substituting a mismatched base in the 3’-end of the reverse primer to form a non-canonical base pair that would further destabilize 3’-end complementarity to a greater degree in templates that lack the ΔY144 deletion, a technique which can be useful for detecting single nucleotide polymorphisms (Gibbs et al., 1989; Lefever et al., 2013; Tsai et al., 1994). These primers (R28-R37) showed the highest specificity with ΔH69/ΔV70 template amplifying over ten cycles later than B.1.1.7 template (Figure 2C). Although some of these reverse primers (R33-35) showed reduced sensitivity when compared to B.1.1.7 template amplified with our common SARS-CoV-2 S gene, we found that judicious placement of an LNA-modified thymine (LNA-T) towards the 3’-end mismatch retained high sensitivity (e.g., R36). This benefit had a limit, however, as placing the LNA-T too close to the mismatch was detrimental to sensitivity (R37; Figure 2D). Taken together, several of these primers are capable of discriminating between B.1.1.7 and other variants containing the ΔH69/ΔV70 deletion, provided that a second reaction is ran in parallel using the SARS-CoV-2 S gene set that can be used as a benchmark to assess the relative sensitivity. If the B.1.1.7 primer set amplifies the sample within five Ct cycles of the SARS-CoV-2 S gene primer set, then the sample is B.1.1.7 positive. Alternatively, if the B.1.1.7 primer set amplifies the sample in 8 or more Ct cycles relative to the SARS-CoV-2 S gene primer set, than the sample likely belongs to a variant that contains the ΔH69/ΔV70 deletion, but not the ΔY144 deletion, and hence is B.1.1.7 negative.
Comparison of B.1.1.7 S gene primer/probe sets
Following the clinical validation, we compared three different versions of B.1.1.7 primer/probe sets that varied according to the reverse primer (V1, V2, and V3 use reverse primers R14, R23, and R36, respectively) using a selected set of 46 samples, some of which where sequenced to confirm lineage status. Given our interpretation criterion (Table 4), we determined that the V3 primer/probe set (F3-P3-P4-R36) performed the best since it correctly identified all B.1.1.7 and ΔH69/ΔV70 deletion samples, with the exception of one ΔH69/ΔV70 deletion sample that was interpreted as inconclusive (Figure 3). While V1 and V2 primer/probe sets correctly identified all B.1.1.7 samples, they performed poorly at calling ΔH69/ΔV70 deletion samples. V1 correctly identified only 11 samples, whereas V2 performed better by correctly identifying 30 samples. Sequencing results on all B.1.1.7 samples and a subset (n=12) of ΔH69/ΔV70 deletion samples confirmed their lineage designation. Among the ΔH69/ΔV70 deletion samples that were sequenced, the V1, V2, and V3 primer/probes sets detected 25%, 67%, and 92%, respectively, of the samples identified as lineage B.1.258, thus confirming V3 as the most suitable primer/probe combination to identify other lineages containing only the ΔH69/ΔV70 deletion.
Analytical sensitivity and clinical evaluation of lineage B.1.1.7 S gene primer/probe set
With our final primer/probe sets for SARS-CoV-2 S gene and B.1.1.7, we multiplexed each assay with the US CDC human RNase P primer/probe set (for sequence, see (Centers for Disease Control and Prevention, 2020)) as an internal control to assess RNA extraction and assay performance. We then assessed the analytical sensitivity using serial dilutions of RNA extracted from a B.1.1.7 positive sample. Both assays displayed high sensitivity (Figure 4A) with our SARS-CoV-2 S gene and B.1.1.7 assays reliably detecting down to only 2 copies/reaction (0.4 copies/μl) and 10 copies/reaction (2 copies/μl), respectively, placing them among the most sensitive SARS-CoV-2 RT-qPCR assays available.
We evaluated the clinical performance of our SARS-CoV-2 S gene and B.1.1.7 assays on 65 clinical samples that underwent sequencing to identify lineage status using interpretation criterion outlined in Table 4. Our SARS-CoV-2 S gene assay detected all 65 clinical samples regardless of lineage (Table 5) confirming its utility as a general screening assay for the most common SARS-CoV-2 variants. Out of 37 clinical samples classified as lineage B.1.1.7 by sequencing, our B.1.1.7 assay positively identified 36 samples, while only one sample was deemed inconclusive. The ΔCt of this sample was slightly greater than five cycles (e.g., Sample 40, ΔCt = 5.7) relative to the Ct value for the SARS-CoV-2 S gene assay, which exceeded our cut-off for a positive identification (Figure 4B).
Notably, our assay was also capable of identifying samples carrying the ΔH69/ΔV70 deletion such as those belonging to the B.1.258 lineage, provided that the sample contains sufficient viral load as other ΔH69/ΔV70 variants yield ΔCt values greater than 8 Ct cycles. For the 16 samples that carry only the ΔH69/ΔV70 deletion and belong to lineage B.1.258, our B.1.1.7 assay correctly identified 13 out of 16 samples. Two samples had relatively high Ct values in the SARS-CoV-2 S gene assay (Sample 33, Ct = 30.1 and Sample 65, Ct = 28.9) and therefore were not detected by the B.1.1.7 assay, making confirmation of the ΔH69/ΔV70 status impossible with our cut-off criterion. One sample had a ΔCt outside the criterion for ΔH69/ΔV70 deletion confirmation and was deemed inconclusive. Detection of human RNase P showed high homogeneity in all analyzed samples, confirming the suitability of this assay as an internal control for collection and RNA extraction from a clinical sample (data not shown). Overall, the clinical evaluation confirmed the diagnostic utility of both our SARS-CoV-2 S gene and B.1.1.7 assays, which showed 100% (65/65) and 97.3% (36/37) diagnostic sensitivity, respectively. For an overview of the clinical evaluation data, lineage of each sample, and GISAID information, see Table 6. We have provided a decision tree (Figure 4C) that users may follow to implement this research use only test to directly detect the presence of the B.1.1.7 variant.
Discussion
The recent emergence of a novel SARS-CoV-2 variant called lineage B.1.1.7 has sparked global consternation as it has now been confirmed in over 70 countries and threatens to exacerbate an already dire pandemic. To mitigate the spread of the B.1.1.7 variant, it is imperative that countries have diagnostic tools that can quickly and accurately detect and track the prevalence of the variant in order to implement the appropriate epidemiological measures. Here we report a novel RT-qPCR test to differentiate the B.1.1.7 variant from other SARS-CoV-2 lineages. The test consists of running two S gene target assays, one specific for B.1.1.7 and the other for all SARS-CoV-2 strains, and performing a simple comparison of relative Ct values that allows the user to differentiate the B.1.1.7 variant from other variants that have the ΔH69/ΔV70 deletion. We validated this test on clinical samples that were sequenced to determine the exact SARS-CoV-2 lineage and the results demonstrated a high level of sensitivity in distinguishing the B.1.1.7. variant. This RT-qPCR test provides a positive identification of the B.1.1.7 variant, providing countries with a powerful tool to detect and track lineage B.1.1.7, especially countries that have considerable prevalence of variants carrying only the ΔH69/ΔV70 deletion (Bal et al., 2021; Brejová et al., 2021; Larsen and Worobey, 2020; Moreno et al., 2021), which are mistakenly identified as B.1.1.7 variants by currently used SGTF assays.
We have provided interested users with the primer and probe sequences to implement this B.1.1.7 assay in their own laboratories with the hope this can rapidly scale the ability of countries to identify the B.1.1.7 variant and implement epidemiological measures to mitigate its spread. This test is also available as a research use only kit called rTest COVID-19 B.1.1.7 qPCR kt (rTest COVID-19 qPCR B.1.1.7 Kit • COVID-19 Diagnostics • MultiplexDX, MultiplexDX, Bratislava, Slovakia) that contains lyophilized SARS-CoV-2 S gene and B.1.1.7 primer/probe sets both multiplexed with human RNase P (200 reactions per assay) and a lyophilized positive control containing B.1.1.7 RNA spiked with human RNA that can be used to validate all primer/probe sets. The kit utilizes the room temperature-stable SOLIScript® 1-step CoV Kit (SolisBiodyne) and laboratory tests show that all kit components are stable for at least one month at room temperature, eliminating cold chain shipping/storage and freeing valuable freezer storage space (e.g., for storage of mRNA-based vaccines). This test can provide labs with a powerful tool to directly confirm the presence of the B.1.1.7 variant in a sample previously determined SARS-CoV-2 positive by an approved screening test, thus avoiding the use of target gene failure assays that can be plagued with low specificity and obviating the need to conduct burdensome and costly genomic sequencing. This is particularly important for countries that are experiencing extensive circulation of variants harboring only the ΔH69/ΔV70 deletion as current RT-qPCR assays that rely on SGTFs erroneously classify these samples as presumptive B.1.1.7 variants.
Please note that the study is presented in a shortened form. A full-length preprint can be found at the following link:
Acknowledgements
We gratefully acknowledge the authors from the submitting and originating laboratories that shared genetic sequencing data with the GISAID initiative. This project was supported by the European Union’s Horizon 2020 research and innovation program [EVA-GLOBAL project, grant agreement number 871029] (BK) and grants from the Slovak Research and Development Agency: PP-COVID-20-0017 (BK) and PP-COVID-20-0116 (PC, BK).
References
Bal, A. et al., 2021. Two-step strategy for the identification of SARS-CoV-2 variant of concern 202012/01 and other variants with spike deletion H69-V70, France, August to December 2020. medRxiv 2020.11.10.20228528. https://doi.org/10.1101/2020.11.10.20228528
Brejová, B. et al., 2021. B.1.258∆, a SARS-CoV-2 variant with ∆H69/∆V70 in the Spike protein circulating in the Czech Republic and Slovakia. (2021).
Centers for Disease Control and Prevention, 2020. Research Use Only 2019-Novel Coronavirus (2019-nCoV) Real-time RT-PCR Primers and Probes [WWW Document]. URL https://www.cdc.gov/coronavirus/2019-ncov/lab/rt-pcr-panel-primer-probes.html (accessed 2.4.21).
Gibbs, R.A., Nguyen, P.N., Caskey, C.T., 1989. Detection of single DNA base differences by competitive oligonucleotide priming. Nucleic Acids Res 17, 2437–2448.
Gouy, M., Guindon, S., Gascuel, O., 2010. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27, 221–224. https://doi.org/10.1093/molbev/msp259
Larsen, B.B., Worobey, M., 2020. Identification of a novel SARS-CoV-2 Spike 69-70 deletion lineage circulating in the United States - SARS-CoV-2 coronavirus / SARS-CoV-2 Molecular Evolution [WWW Document]. Virological. URL https://pando.tools/t/identification-of-a-novel-sars-cov-2-spike-69-70-deletion-lineage-circulating-in-the-united-states/577 (accessed 1.29.21).
Latorra, D., Arar, K., Michael Hurley, J., 2003. Design considerations and effects of LNA in PCR primers. Molecular and Cellular Probes 17, 253–259. Redirecting
Lefever, S. et al., 2013. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem 59, 1470–1480. https://doi.org/10.1373/clinchem.2013.203653
Levin, J.D. et al., 2006. Position-dependent effects of locked nucleic acid (LNA) on DNA sequencing and PCR primers. Nucleic Acids Res 34, e142–e142. https://doi.org/10.1093/nar/gkl756
McTigue, P.M., Peterson, R.J., Kahn, J.D., 2004. Sequence-Dependent Thermodynamic Parameters for Locked Nucleic Acid (LNA)−DNA Duplex Formation. Biochemistry 43, 5388–5405. https://doi.org/10.1021/bi035976d
Moreno, G. et al., 2021. Detection of non-B.1.1.7 Spike ∆69/70 sequences (B.1.375) in the United States - SARS-CoV-2 coronavirus / nCoV-2019 Genomic Epidemiology [WWW Document]. Virological. URL https://pando.tools/t/detection-of-non-b-1-1-7-spike-69-70-sequences-b-1-375-in-the-united-states/587 (accessed 2.4.21).
Nagy, A et al., 2017. Evaluation of TaqMan qPCR System Integrating Two Identically Labelled Hydrolysis Probes in Single Assay. Scientific Reports 7, 41392. Evaluation of TaqMan qPCR System Integrating Two Identically Labelled Hydrolysis Probes in Single Assay | Scientific Reports
Nakamura, T. et al. , 2018. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34, 2490–2492. https://doi.org/10.1093/bioinformatics/bty121
Resende, P.C. et al., 2020. SARS-CoV-2 genomes recovered by long amplicon tiling multiplex approach using nanopore sequencing and applicable to other sequencing platforms. bioRxiv 2020.04.30.069039. https://doi.org/10.1101/2020.04.30.069039
Tsai, M.Y. et al., 1994. Determination of a T/G polymorphism at nucleotide 3206 of the apolipoprotein C III gene by amplification refractory mutation system. Clin Chem 40, 2235–2239.
Yip, S.P. et al., 2005. Use of Dual TaqMan Probes to Increase the Sensitivity of 1-Step Quantitative Reverse Transcription-PCR: Application to the Detection of SARS Coronavirus. Clin Chem 51, 1885–1888. https://doi.org/10.1373/clinchem.2005.054106
Zuker, M., 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31, 3406–3415. https://doi.org/10.1093/nar/gkg595
Table 1. Origin and genomic characterization of GISAID sequences used for alignment and primer/probe design.
Table 2. Oligonucleotide primers and probes for common SARS-CoV-2 S gene primer/probe set.

Table 3. Oligonucleotide primers and probes for lineage B.1.1.7 S gene primer/probe set

Table 4. Interpretation of SARS-CoV-2 test results and corresponding actions
| SARS-CoV-2 S gene | ΔCt between SARS-CoV-2 S gene and B.1.1.7 | Human RNase P | Result interpretation | Report |
|---|---|---|---|---|
| + | Max 5 Ct | +/ND | SARS-CoV-2 B.1.1.7 detected | SARS-CoV-2 B.1.1.7 positive |
| + | Min 8 Ct | +/ND | SARS-CoV-2 ΔH69/ΔV70 deletion detected | SARS-CoV-2 B.1.1.7 negative |
| + | Min 20 Ct | +/ND | Other lineage of SARS-CoV-2 detected | SARS-CoV-2 B.1.1.7 negative |
| + | ND | +/ND | Consensus or other lineage of SARS-CoV-2 detected | SARS-CoV-2 B.1.1.7 negative |
| ND | + | +/ND | Inconclusive result | Inconclusive |
| ND | ND | ND | Invalid result | Invalid |
ND, not detected
Table 5. Clinical performance of SARS-CoV-2 S gene and B.1.1.7 primer/probes sets
| Analyzed samples | Sequencing | SARS-CoV-2 S gene | B.1.1.7 (ΔH69/ΔV70 + ΔY144) | ΔH69/ΔV70 only | Other lineage of SARS-CoV-2 | Inconclusive |
|---|---|---|---|---|---|---|
| B.1.1.7 (ΔH69/ΔV70 + ΔY144) | 37 | 37 | 36 | 0 | 0 | 1 |
| ΔH69/ΔV70 only | 16 | 16 | 0 | 13 | 2 | 1 |
| Other lineage of SARS-CoV-2 | 12 | 12 | 0 | 0 | 12 | 0 |
Table 6. Overview of clinical sample RT-qPCR results, lineage, and GISAID information

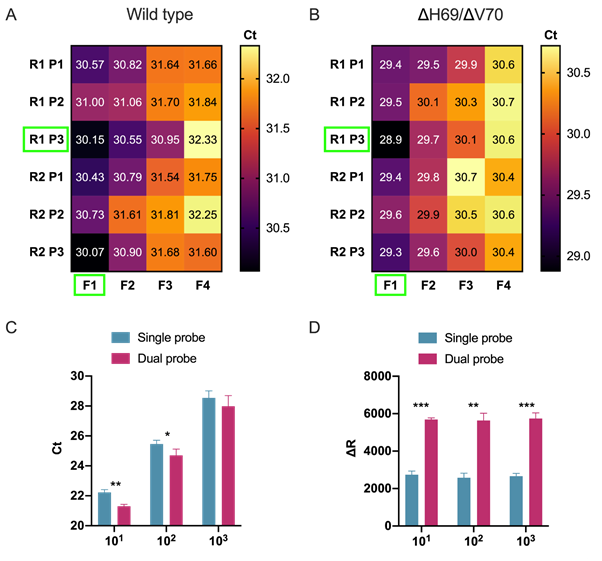
Figure 1. Development and optimization of a general SARS-CoV-2 S gene primer/probe set for all SARS-CoV-2 variants .
A, B) Heatmaps illustrate oligonucleotide primer and probe combinations designed to target conserved sequences within the spike gene, including all SARS-CoV-2 variants that were contained in our bioinformatics analysis. Combinations of forward (F1-F4) and reverse primers (R1-R2) and hydrolysis probes (P1-P2) were tested using two separate SARS-CoV-2 variants, a common SARS-CoV-2 variant (Wild type, panel A) and a variant containing the ΔH69/ΔV70 deletion (B). Green rectangle boxes indicate best performing primer/probe combinations. C, D) Bargraphs compare RT-qPCR performance of a single probe versus an additional identically labelled dual probe using three 10-fold (101, 102, 103) dilutions of SARS-CoV-2 template ran in triplicates. Evaluation of the performance was done by comparing raw Ct values (C) and fluorescence intensity values (D). Statistical analysis was performed using paired t-test (***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05).
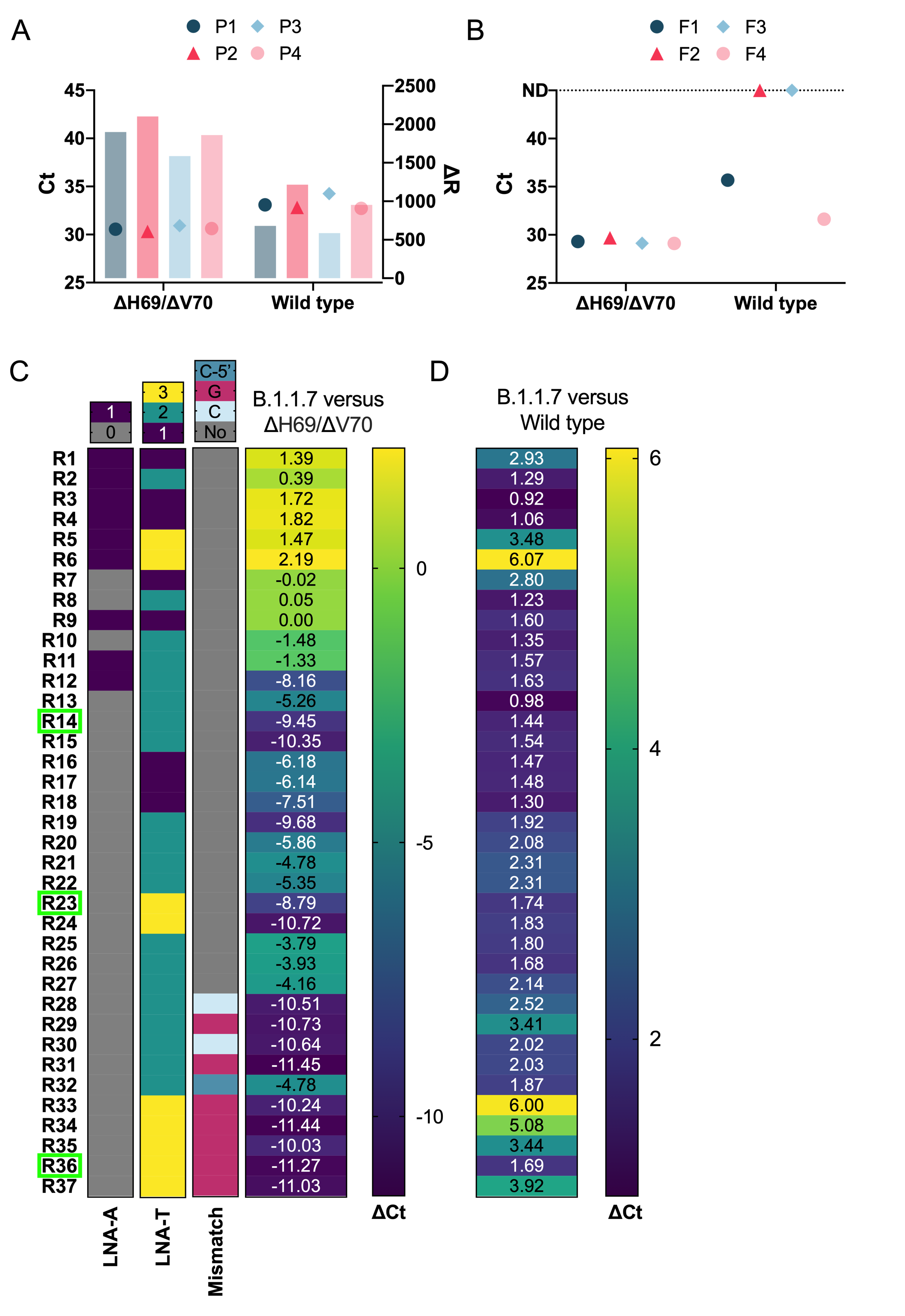
Figure 2. Development and optimization of a spike gene primer/probe set specific for the B.1.1.7 SARS-CoV-2 variant .
A) Comparison of probes (P1-4) targeting the ΔH69/ΔV70 deletion in B.1.1.7 using the best forward and reverse primers from the SARS-CoV-2 S gene primer/probe set (from Figure 1/Table 2). The performance between ΔH69/ΔV70 deletion template and wild type template is depicted by comparing Ct values (colored symbols) and fluorescence intensity (ΔR, colored bars). B) Assessment of forward primers (F1-4) targeting the ΔH69/ΔV70 deletion in B.1.1.7 using the best reverse primer and probe (from Figure 1/Table 2). Symbols compare Ct values of the ΔH69/ΔV70 variant and wild type templates. Dotted line indicates samples that were not detected (ND) within 45 cycles. C) Overview of reverse primer designs targeting the ΔY144 deletion of the B.1.1.7 variant and their effects on specificity by comparing the relative ΔCt when amplifying either B.1.1.7 or ΔH69/ΔV70 variants as template. Darker colors in the heatmap represent a greater ΔCt and consequently better specificity. Green rectangle boxes indicate reverse primers selected for further optimization. LNA-A depicts primers containing an LNA modified adenine base located at either the 3’- or 5’-end of the reverse primer. LNA-T displays the number (1-3) of LNA-modified thymine bases for each reverse primer. Mismatch base represents design modifications to introduce either a guanine (G) or cytosine (C) mismatch base in either the penultimate base (G or C) or the 3rd from last base (C-5’) relative to the 3’-end of the reverse primer. D) Heatmap shows ΔCt value comparison of B.1.1.7 primer/probe set to SARS-CoV-2 S gene primer/probe set using the B.1.1.7 variant as template. Darker colors indicate smaller ΔCt and consequently better specificity.
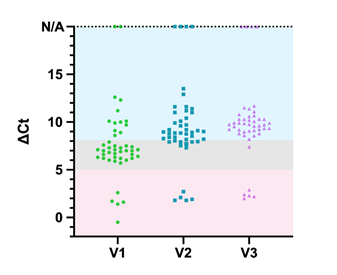
Figure 3. Overview of B.1.1.7 assay performance on clinical samples.
Three different versions (V1, V2, V3) of primer/probe sets for detection of the B.1.1.7 variant were directly compared on a selected panel of 46 SARS-CoV-2 positive clinical samples, some of which were confirmed B.1.1.7 and B.1.258 variants by sequencing. ΔCt values correspond to SARS-CoV-2 S gene assay Ct – B.1.1.7 assay Ct. Colored boxes within the plot define boundaries for corresponding variant interpretation, red (ΔCt ±5) for B.1.1.7, blue (ΔCt 8-20) for ΔH69/ΔV70, grey (ΔCt 5-8) for inconclusive samples. N/A represents samples which were detected only in SARS-CoV-2 S gene assay and therefore are interpreted as consensus SARS-CoV-2.

Figure 4. Analytical sensitivity and clinical validation of SARS-CoV-2 S gene and B.1.1.7 assays.
A) The limit of detection was determined for both SARS-CoV-2 S gene and B.1.1.7 assays by serial dilutions of isolated viral B.1.1.7 RNA. Data depict the mean and SD of eight replicates per each dilution. The dotted line at Ct 40 serves as a threshold after which amplification is considered invalid. B) Overview of ΔCt values (= SARS-CoV-2 S gene assay Ct. – B.1.1.7 assay Ct) for each sample in the clinical validation. Symbols represent the various SARS-CoV-2 lineages that were identified by sequencing. Closed symbols represent samples correctly identified by either the SARS-CoV-2 S gene or B.1.1.7 assays, whereas open symbols denote samples that did not meet the criterion established for variant identification. The shaded background shows ΔCt ranges that correspond with the criterion to report a sample as B.1.1.7 positive (pink), ΔH69/ΔV70 deletion positive (teal), and inconclusive (gray). ND, not detected. C) Decision tree demonstrating the proper workflow, interpretation criterion, and actions to implement the SARS-CoV-2 S gene and B.1.1.7 assays a testing regime to identify B.1.1.7 positive samples.