Key points
- We generated 64 new Lassa virus genomes from clinical samples collected at Irrua Specialist Teaching Hospital from 2015-2016,
- A phylogenetic analysis of these sequences reveals clustering according to broad geographic regions in Nigeria: southwestern, eastern, and northern, separated by major rivers. These rivers likely serve as ecological barriers of the Mastomys rodent.
- The phylogenetic and geographical data suggest multiple independent cross-species transmission events from the reservoir to humans, rather than human-to-human transmission.
- This data will inform ongoing analysis of Lassa fever in 2018.
Lassa fever (LF) incidence has increased over the last 3 years in Nigeria. Lassa virus (LASV), the etiologic agent of LF and Lassa hemorrhagic fever, is enzootic in Mastomys rodents in West Africa. Most human LASV infections result from reservoir-to-human transmission, yet the potential for sustained human-to-human transmission, which has occurred in hospital settings, remains a focus of public health monitoring. LASV sequencing is a useful method to determine viral diversity and monitor transmission patterns. Previous genetic sequencing has revealed the ancient origins of LASV as well its extensive diversity, especially in Nigeria, with 4 separate deep lineages (Andersen et al. 2015). However, little is known about LASV diversity since 2014.
To assist in the understanding of the current increase in cases, we are publicly releasing 64 LASV genomes from confirmed clinical samples from 2015 and 2016. All samples were received and diagnosed at Irrua Specialist Teaching Hospital (ISTH). The majority of samples are from the states of Ondo and Edo, where ISTH is located, with additional samples referred to ISTH from elsewhere in Nigeria for diagnostics purposes. Samples were prepared and sequenced at Redeemer’s University (Ede, Nigeria) or the Broad Institute (Cambridge, MA, USA) using Illumina MiSeq and HiSeq 2000 technologies and analyzed using our publicly available software viral-ngs (GitHub - broadinstitute/viral-ngs: Viral genomics analysis pipelines) implemented on the DNAnexus cloud compute platform.
Within these 64 historical samples from 2015-2016, we observe geographical and epidemiological barriers to transmission and no substantial evidence for extensive human-to-human transmission of LASV. Phylogenetic analysis of the LASV genome S segment reveals clustering according to broad geographic regions in Nigeria (Fig. 1A). In addition to the previously described lineage III that is concentrated in northern Nigeria, we observe sub-lineage within lineage II comprising samples from the eastern states of Ebonyi, Taraba and Anambra. These regions, and LASV lineages therein, are separated by major rivers that could provide substantial biogeographic barriers to the movement of the Mastomys rodent (Fig. 1B). The long branch lengths of these groups further suggests the existence of extensive diversity of circulating LASV in Nigeria that has yet to be fully captured. The absence of extensive clustering or a ‘ladder-like’ structure from the phylogeny of the new sequences suggests multiple independent cross-species transmission events, rather than human-to-human transmission.
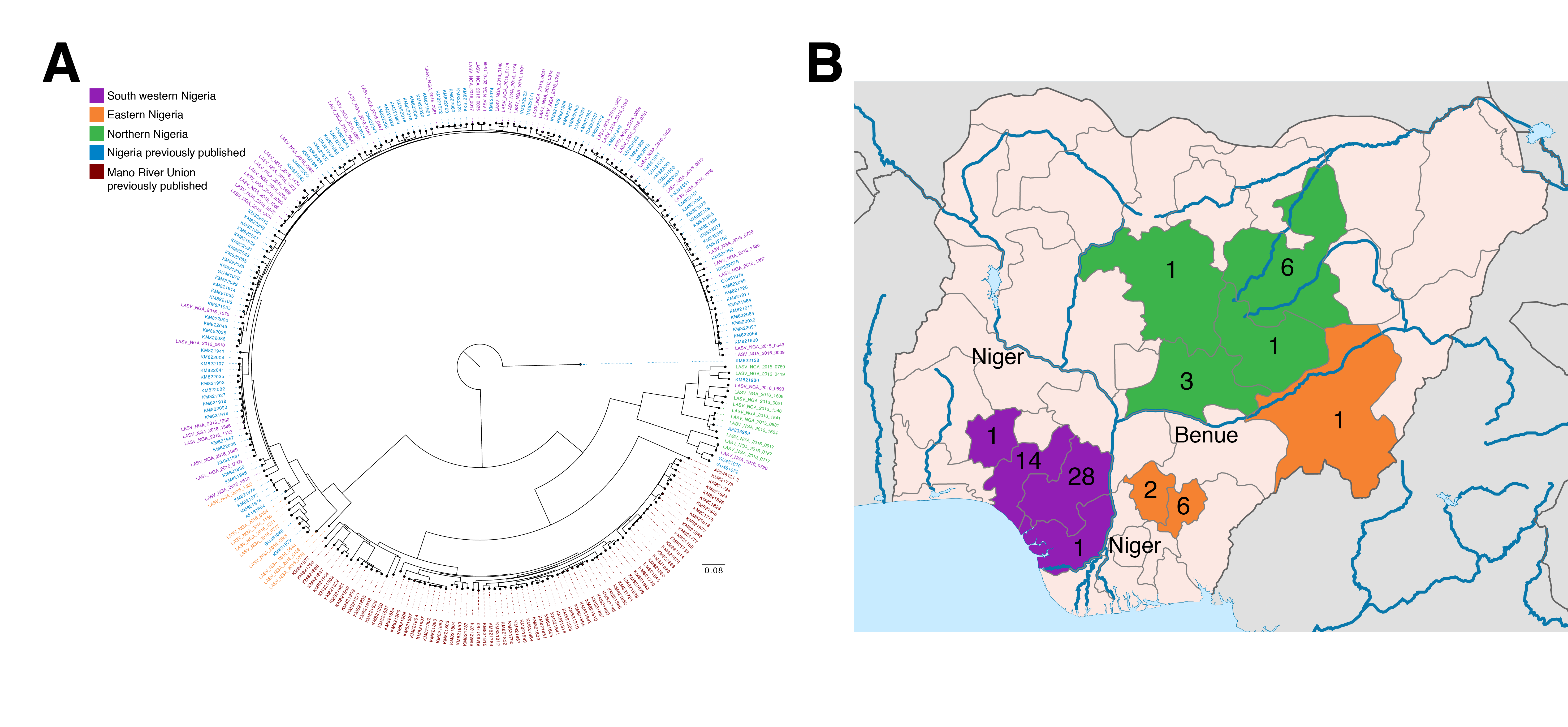
Figure 1. Distribution of LASV genome diversity in Nigeria. A) Phylogenetic tree of the S segment of the Lassa virus genome. The 64 newly generated samples are colored by region (north (green), east (orange) and south west (purple)) and shown alongside previously published samples from Nigeria (blue) and the Mano River Union (red). Mano River Union samples are predominantly from Sierra Leone. The tree is rooted using the Pinneo strain. B) Map showing the numbers of samples included in the current dataset by state. Colours are as in A. Major rivers are marked in blue and named. B) Map is adapted from a map by Ali Zifan (Own work) [CC BY-SA 4.0 (Creative Commons — Attribution-ShareAlike 4.0 International — CC BY-SA 4.0)], via Wikimedia Commons.
These preliminary results raise important questions about the current increase in cases and highlight the urgent need to sequence more samples from 2018 to determine if this trend continues to present. We are currently engaged in this work, to sequence all LASV cases from 2017 and 2018 to understand the genetic diversity of LASV strains in recent samples to help inform public health efforts.
Data availablitiy
Genome sequences for all samples are available here:
https://storage.googleapis.com/sabeti-public/data_release/lasv_nga/round1/lasv-acegid-round1-fasta_only.tar.gz
https://storage.googleapis.com/sabeti-public/data_release/lasv_nga/round1/lasv-acegid-round1-gbf_only.tar.gz
Partners and collaborators
This work is part of ongoing research collaborations between several partners:
Irrua Specialist Teaching Hospital and Institute for Lassa Fever Research and Control (Irrua, Nigeria)
African Center of Excellence for Genomics of Infectious Disease (ACEGID Redeemer’s University, Ede, Nigeria)
Center for Viral Systems Biology (La Jolla, CA, USA)
Joint West Africa Research Group (Walter Reed Army Institute of Research, Bethesda, MD, USA)
Viral Hemorrhagic Fever Consortium (New Orleans, LA, USA)
Broad Institute of MIT and Harvard (Cambridge, MA, USA)
Harvard University (Cambridge, MA, USA)
Statement on continuing work and analyses prior to publication
These genomes are being shared pre-publication to help the research and public health communities respond to the current increase in Lassa cases. We encourage others to download, share, use, and analyze this data. Please note though that this data is still based on work in progress and should be considered preliminary. Our analyses of this data is ongoing and a publication communicating our findings on these and other published genomes is in preparation. If you intend to use these sequences prior to our publication, please contact us directly to coordinate. We will continue to release LASV genomes, prior to publication, as soon as they are generated to update the community.
Professor Christian Happi (Redeemer’s University, ACEGID): [email protected]
Professor Pardis Sabeti (Harvard University, Broad Institute): [email protected]
Dr Katherine Siddle (Harvard University, Broad Institute): [email protected]
References
Andersen, K. G., B. J. Shapiro, C. B. Matranga, R. Sealfon, A. E. Lin, L. M. Moses, O. A. Folarin, et al. 2015. “Clinical Sequencing Uncovers Origins and Evolution of Lassa Virus.” Cell 162: 738–50.