To date, the closest described relative to the nCoV-2019 novel coronavirus in a non-human host is a bat SARSr-CoV called RaTG13 (genbank accession MN996532, Zhou et al (2020) [1]) sampled from a Rhinolophus affinis bat in Yunnan Province in 2013.
Although this has a 96.1% identity with nCoV-2019, at the rate that coronaviruses evolve, this represents signifiant evolutionary time. This can be estimated using BEAST [3,4] by assuming a rate of evolution. Here I have show estimates for a rate of 1e-3 and 5e-4 substitutions per site per year. The former rate seems to be close to that observed in the human outbreak and the latter closer to rates estimated for some other coronaviruses.
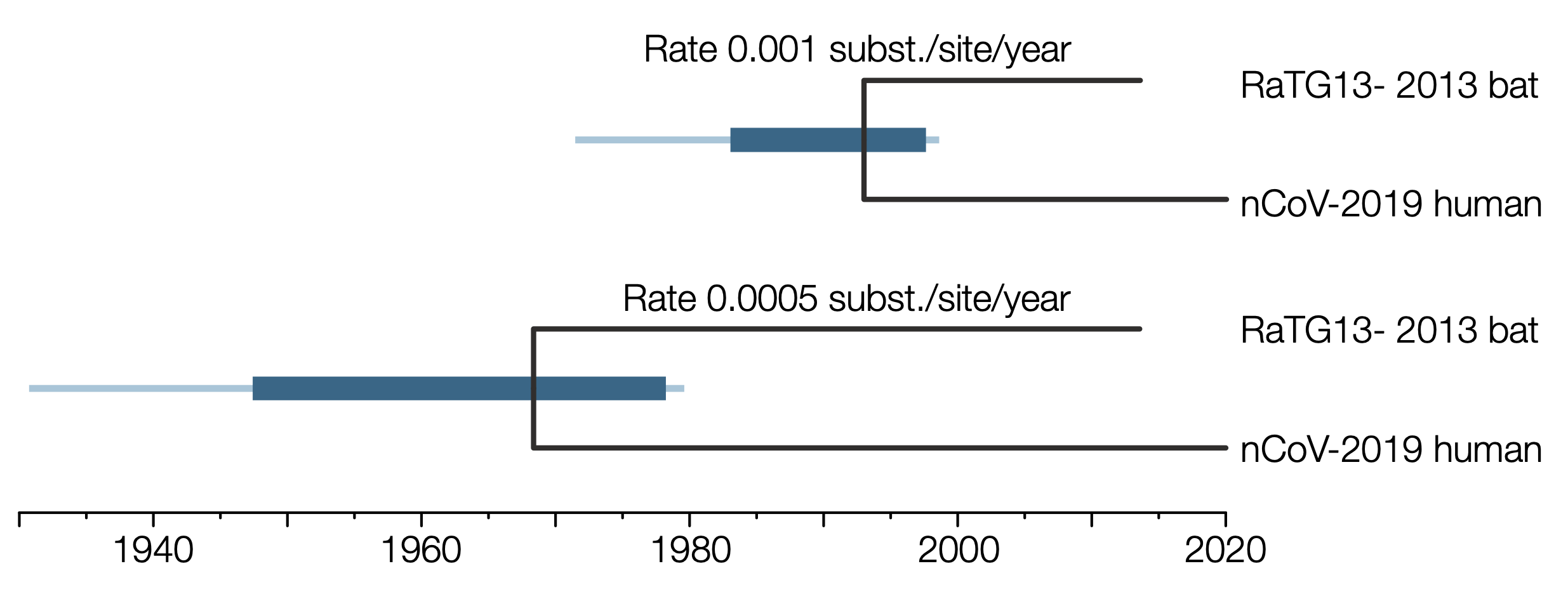
Figure 1 | The divergence time estimate between nCoV-2019 and RaTG13 SARSr-CoV. Bars represent the 95% HPD credible intervals, lines represent the complete range of sampled values. Analysis in BEAST v1.10.4 with GTR+G8 substitution model, strict molecular clock, constant size population coalescent prior.
With the faster rate this analysis suggests the human nCoV-2019 and the bat SARSr-CoV last shared a common ancestor around the end of 1992 and likely before mid-1997 (the upper 95% credible interval). For the slower rate this estimate is proportionally older — likely before 1978.
Note — this is an estimate of when the ancestors of RaTG13 and nCoV-2019 were in the same host individual (very likely a bat). It is certainly not when it jumped into humans (that was likely very shortly before the earliest recorded cases in December 2019 - see this post).
Recently it has been announced that a coronavirus recovered from a pangolin may have 99% identity to nCoV-2019 making this a far closer relative. Assuming the faster rate of evolution this would imply a most recent common ancestor approximately 6 years ago (2014).
Addendum — as @R_H_Ebright pointed out on Twitter the exact phrase used was “as high as 99%”.
| virus | isolate | host | accession | reference | |
|---|---|---|---|---|---|
| SARSr-CoV | RaTG13 | Rhinolophus affinis | MN996532 | Zhou et al (2020) | [1] |
| nCoV-2019 | Wuhan-Hu-1 | human | NC_045512 | Wu et al (2020) | [2] |
Table 1 | virus genome sequence used in this analysis.
References
- Zhou, P., Yang, X., Wang, X. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin | Nature
- Wu, F., Zhao, S., Yu, B. et al. A new coronavirus associated with human respiratory disease in China. Nature (2020). A new coronavirus associated with human respiratory disease in China | Nature
- Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol (2012) 29: 1969–1973.
- Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol (2018) 4: vey016.
