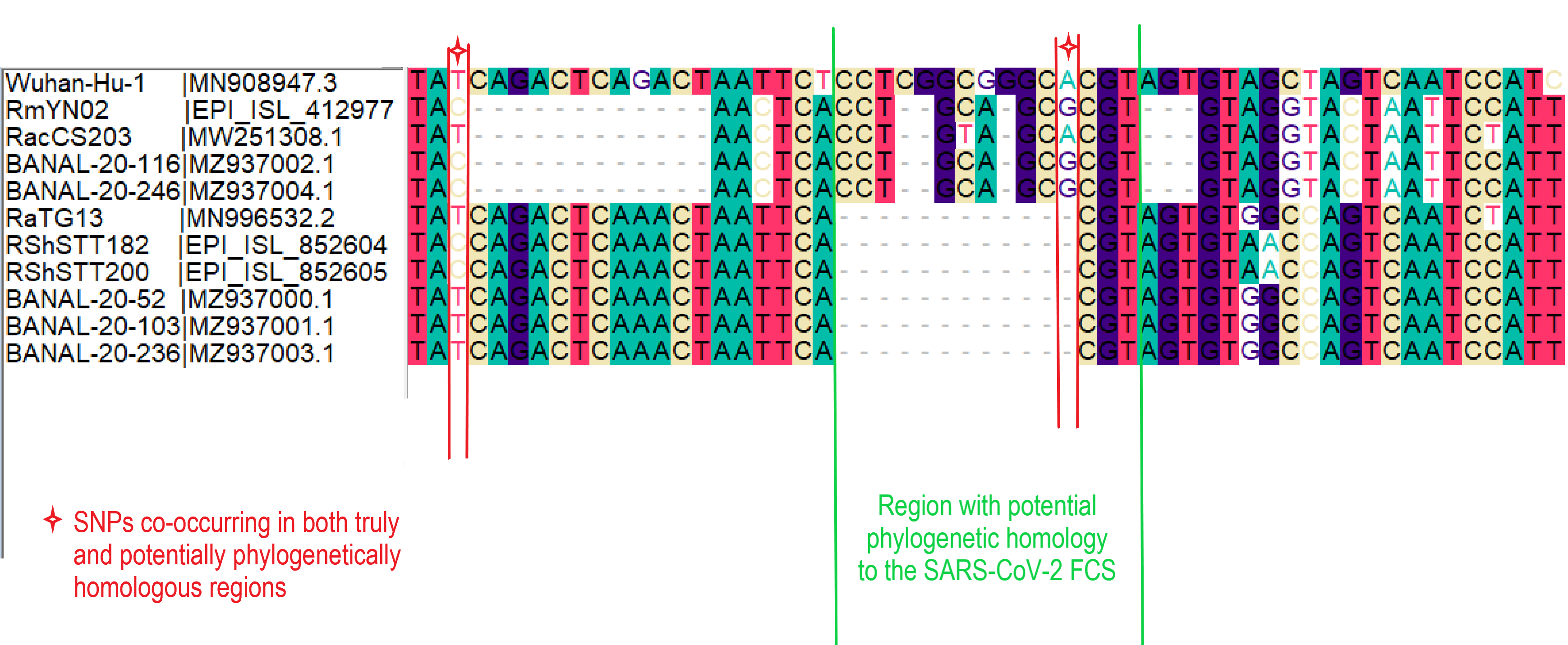
Sequencing the SARS-CoV-2 genome has massively aided the effort to understand and contribute to the control of the COVID-19 pandemic, through comparative genetics and molecular epidemiology. However, due to the very limited sampling of closely related non-human coronaviruses, the origin of some genomic regions remains an open question. One region that has probably attracted the most interest and speculation is the polybasic furin cleavage site insertion in the Spike open-reading frame of SARS-CoV-2, absent from all closely related Sarbecoviruses sampled to date (Andersen et al., 2020).
The most notable effort to identify the origin of this furin site has been made by William Gallaher (2020), suggesting a copy-choice recombination error between the proximal ancestor of SARS-CoV-2 and a yet unsampled betacoronavirus. This is based on detectable sequence homology between the SARS-CoV-2 oligomer insert and a downstream region of another bat coronavirus genome, HKU9-1. Could this recombination event have occurred within the SARS-CoV-2 Sarbecovirus lineage? While investigating the sequence similarity between the SARS-CoV-2 genome and the newly sampled RmYN02 sequence (Zhou et al., 2020) we identified an overlooked fragment of sequence homology with the SARS-CoV-2 furin site, providing a clue about the likely sequence this recombined from (MacLean/Lytras et al., 2020).
The RmYN02 recombinant region
RmYN02, although a recombinant, is the closest known relative of SARS-CoV-2 for the majority of its genome (Zhou et al., 2020), with an estimated divergence in the 1970s (MacLean/Lytras et al., 2020). Interestingly, RmYN02’s genome has a long region encompassing the first half of the Spike ORF (Zhou et al., 2020), acquired by recombination from a currently unsampled viral lineage (we designate clade X). Part of this recombinant region is where the sequence corresponding to the SARS-CoV-2 furin site is (Figure 1). Considering the period of time (decades) since RmYN02 and SARS-CoV-2 shared a common ancestor - and the clear recombination evidence between RmYN02 and unknown clade X virus - these viruses must have co-circulated in the bats in the same geographical location, and occasionally co-infected the same individuals.
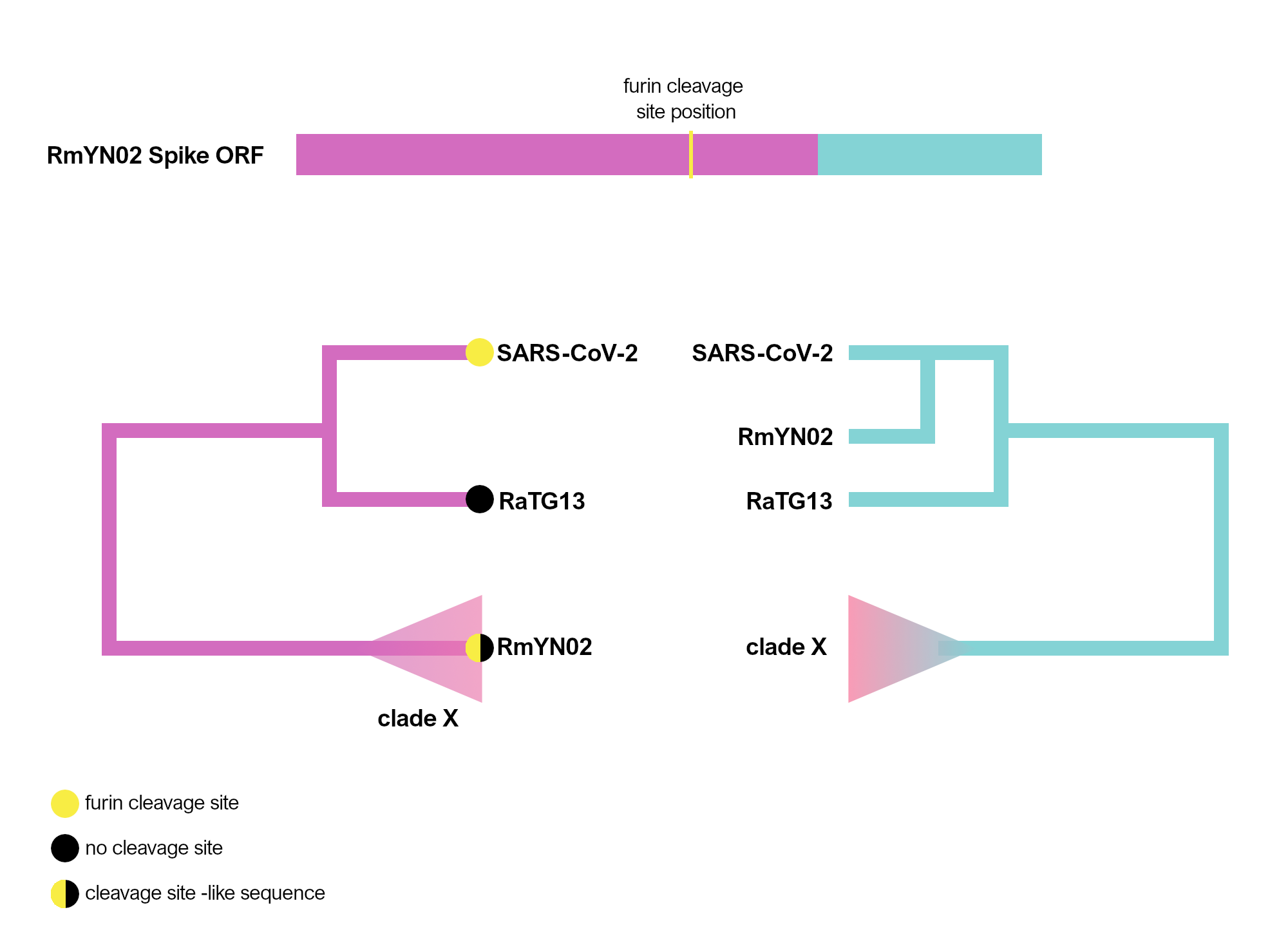
Figure 1. Schematic of the RmYN02 Spike open-reading frame (top) with recombinant regions highlighted in magenta and blue. The position of the furin site is annotated in yellow. The alternative evolutionary histories are shown for each sequence region (bottom). Tip labels on the 5’ tree represent the presence/absence of the furin-like sequence. The hypothetical clade X is shown.
When aligning the furin site region between RmYN02 and SARS-CoV-2, most alignment algorithms would show the furin site sequence as the inserted region (Figure 2A). However, there is clear nucleotide sequence identity between the RmYN02 sequence (AACTCACCTGCAGCG) and the SARS-CoV-2 furin site region, despite the larger number of gaps under this alignment (Figure 2B). Given that the unsampled clade X viruses recombined with SARS-CoV-2’s sister lineage, RmYN02, we propose that the furin site insertion was acquired from a clade X virus when co-infecting the same individual. This would indicate the recombination event giving rise to the important furin cleavage site probably arose in a co-infected bat host, although we can’t discount this taking place in a co-infected intermediate species or even human host (MacLean/Lytras et al., 2020).
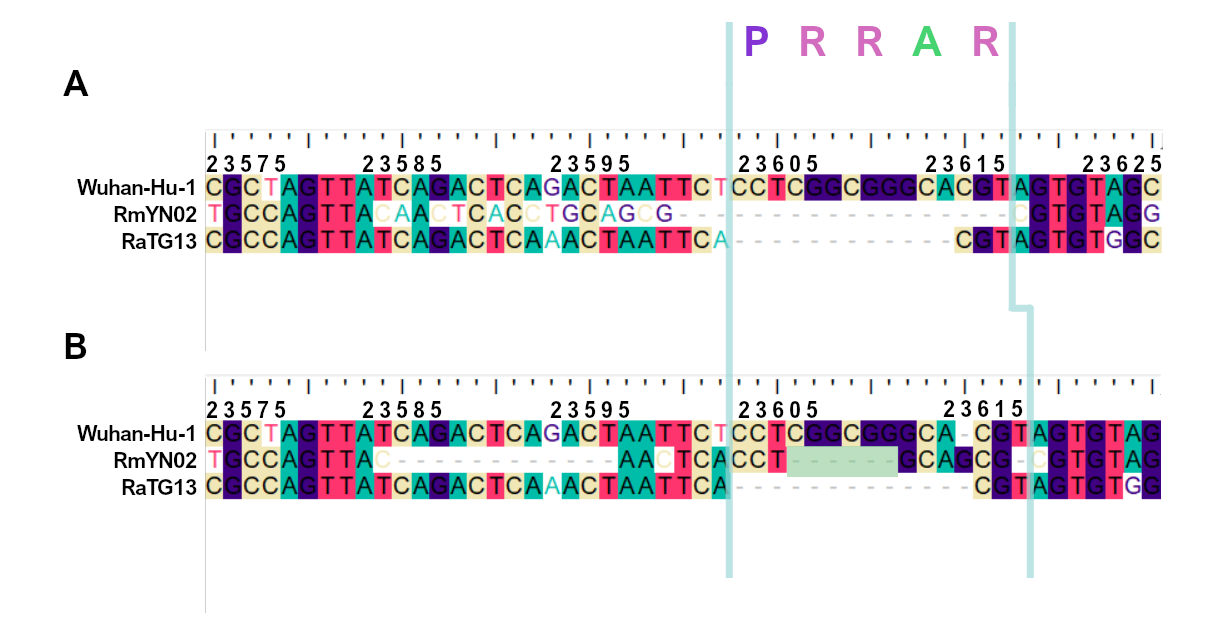
Figure 2. Alternative alignments of the SARS-CoV-2 reference sequence (Wuhan-Hu-1) with RmYN02 at the furin site region. The low sequence identity from positions 23,585 to 23,599 between Wuhan-Hu-1 and RmYN02 (panel A), relative to the alternative alignment (panel B), shows there’s a homologous region to the furin cleavage site present in RmYN02 (positions 23603 to 23615). This indicates the inserted region in the SARS-CoV-2 progenitor was acquired by recombination in a host co-infected with divergent Sarbecoviruses. RaTG13 is also included for clarity. Nucleotide coordinates are mapped to the Wuhan-Hu-1 whole genome sequence. The putative deletion of nucleotides in RmYN02 are highlighted in green.
Coronavirus template switching
Coronaviruses have a more sophisticated polymerase complex than most RNA viruses, and utilise RNA folding to maintain their very large genome size (Sola et al., 2015). Recombination patterns among coronaviruses and Sarbecoviruses in particular, are evident and abundant when examining the genomes of sampled viruses (Boni et al., 2020). Yet, the exact mechanisms through which these frequent recombination events take place between Betacoronaviruses have had little mention in the recent SARS-CoV-2 literature.
A hint to the molecular mechanism responsible for the furin insertion is a previously documented phenomenon in many coronaviruses called ‘leader-switching’. This is when a ‘leaderless’ defective interfering coronaviral RNA (lacking the short sequence required for transcription regulation) acquires a leader sequence from a heterologous coronavirus in a cell during negative RNA synthesis (Chang, Krishnan and Brian, 1996; Stirrups et al., 2000; Ke, Liao and Wu, 2013). We propose that a similar polymerase crossover event produced the insertion in a bat co-infection with the proximal SARS-CoV-2 ancestor and a clade X virus (Animation 1).
The palindromic sequence right before the furin insertion in SARS-CoV-2 (CAGACTCAGACT) is a very likely culprit for why this template switch event happened (Gallaher, 2020). When the helicase encountered this folded RNA tandem repeat, the polymerase temporarily dissociated from the pre-SARS-CoV-2 template, continuing negative RNA synthesis at the homologous coordinates of the clade X genome template co-infecting the cell. The two single nucleotide gaps on the 3’ of the furin site in each viral sequence can be explained by slippage when the polymerase switched to the heterologous sequence, i.e., continuing synthesis from the A (position 23614 corresponding to the SARS-CoV-2 reference, Figure 2). Six in-frame nucleotides encoding for two arginines (CGG CGG) are also missing from the RmYN02 genome (Figure 2B). These have presumably been deleted in RmYN02 (or one of its ancestors) since the recombination event with the clade X virus or represent polymorphisms between the clade X strain that recombined with the RmYN02 ancestor and the one possessing the furin site -like template sequence.

Animation 1. Frame 1: polymerase synthesises negative stranded RNA on the pre-furin SARS-CoV-2 ancestor template sequence (pre-SARS-CoV-2); Frame 2: helicase reaches the folded palindromic sequence; Frame 3: polymerase dissociates and binds to the co-infecting clade X virus template; Frame 4: polymerase continuous RNA synthesis on the clade X template starting from the adenine; Frame 5: polymerase dissociates again and returns to the pre-SARS-CoV-2 template; Frame 6: RNA synthesis continuous on the pre-SARS-CoV-2 template; Frame 7: sequence similarity of SARS-CoV-2 with putative synthesised RNA sequence; Frame 8: furin site protein sequence. The pre-SARS-CoV-2 and clade X virus sequence have been manually inferred (see Methods).
A likely hypothesis
We are unlikely to determine the exact molecular event responsible for the furin cleavage site insertion in SARS-CoV-2. However, as we demonstrate here, we should be able to infer the process that took place. We believe the hypothesis presented here is the most likely explanation so far, primarily supported by the strong assumption that the clade X viruses co-circulated with the proximal ancestors of both RmYN02 and SARS-CoV-2 after the two lineages separated. This hypothesis is also consistent with the complex recombination mechanisms that govern coronaviral diversity (Graham and Baric, 2010; Dudas and Rambaut, 2016), fully supporting a natural origin of the furin cleavage site insertion.
Methods
Spike nucleotide alignments for SARS-CoV-2, RmYN02 and RaTG13 were created using MAFFT (Katoh and Standley, 2013). Alignments were later edited on Bioedit. Animation 1 presents the putative ancestral sequences of pre-SARS-CoV-2 and the clade X virus involved in the putative template switch event. RaTG13 (which possesses the closest sampled sequence for the furin site region to that of SARS-CoV-2) and SARS-CoV-2 were used to infer the pre-SARS-CoV-2 sequence, by retaining identical nucleotides and representing differences in the sequence using IUPAC ambiguity codes, i.e. Y: C or T, W: A or T, R: A or G. The insertion sequence was also excluded in the pre-SARS-CoV-2 sequence. The clade X virus sequence was based almost entirely on the RmYN02 sequence with the exception that the two arginine codons (CGG CGG) on the insertion region were added in accordance to the current SARS-CoV-2 sequence. The alignment of existing and reconstructed sequences is attached below (anc_recon_al.fas).
Spyros Lytras, Oscar A. MacLean, David L. Robertson
MRC-University of Glasgow Centre for Virus Research, Scotland, UK.
References
Andersen, K. G. et al. (2020) ‘The proximal origin of SARS-CoV-2’, Nature Medicine . Nature Research, pp. 450–452. doi: 10.1038/s41591-020-0820-9.
Boni, M. F. et al. (2020) ‘Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic.’, Nature microbiology . Nature Publishing Group, pp. 1–10. doi: 10.1038/s41564-020-0771-4.
Chang, R.-Y., Krishnan, R. and Brian, D. A. (1996) The UCUAAAC Promoter Motif Is Not Required for High-Frequency Leader Recombination in Bovine Coronavirus Defective Interfering RNA , JOURNAL OF VIROLOGY .
Dudas, G. and Rambaut, A. (2016) ‘MERS-CoV recombination: implications about the reservoir and potential for adaptation’, Virus Evolution , 2(1), p. vev023. doi: 10.1093/ve/vev023.
Gallaher, W. R. (2020) ‘A palindromic RNA sequence as a common breakpoint contributor to copy-choice recombination in SARS-COV-2’, Archives of Virology . Springer, 1, p. 3. doi: 10.1007/s00705-020-04750-z.
Graham, R. L. and Baric, R. S. (2010) ‘Recombination, Reservoirs, and the Modular Spike: Mechanisms of Coronavirus Cross-Species Transmission’, Journal of Virology . American Society for Microbiology, 84(7), pp. 3134–3146. doi: 10.1128/jvi.01394-09.
Katoh, K. and Standley, D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution , 30(4), pp. 772–780. doi: 10.1093/molbev/mst010.
Ke, T.-Y., Liao, W.-Y. and Wu, H.-Y. (2013) ‘A Leaderless Genome Identified during Persistent Bovine Coronavirus Infection Is Associated with Attenuation of Gene Expression’, PLoS ONE . Edited by V. Thiel. Public Library of Science, 8(12), p. e82176. doi: 10.1371/journal.pone.0082176.
MacLean, O. A./Lytras S. et al. (2020) ‘Natural selection in the evolution of SARS-CoV-2 in bats, not humans, created a highly capable human pathogen’, bioRxiv . Cold Spring Harbor Laboratory, p. 2020.05.28.122366. doi: 10.1101/2020.05.28.122366.
Sola, I. et al. (2015) ‘Continuous and Discontinuous RNA Synthesis in Coronaviruses’, Annual Review of Virology . Annual Reviews, 2(1), pp. 265–288. doi: 10.1146/annurev-virology-100114-055218.
Stirrups, K. et al. (2000) ‘Leader switching occurs during the rescue of defective RNAs by heterologous strains of the coronavirus infectious bronchitis virus’, Journal of General Virology . Society for General Microbiology, 81(3), pp. 791–801. doi: 10.1099/0022-1317-81-3-791.
Zhou, H. et al. (2020) ‘A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein’, Current Biology . doi: 10.1016/j.cub.2020.05.023.