We are performing daily screens of individual genes from the SARS-CoV-2 genomes from GISAID to keep track of variants that may be positively or negatively selected. This analysis is for the current human pandemic viruses (it does not consider selection during zoonosis or in other species; there are plenty other studies that have done it).
Given the rate of sequencing, this is a unique opportunity to see if any of these analyses have utility in identifying potentially interesting sites; whereas traditionally they are used to examine evolution retrospectively.
At the moment it appears that (as expected) there is not a whole lot going on, with many “high diversity” sites like S D614 not showing much signal for positive selection. There are few potential interesting sites where frequencies are increasing over time and sequences with variants are not in a single tree clade (see http://covid19.datamonkey.org and https://observablehq.com/@spond/natural-selection-analysis-of-sars-cov-2-covid-19), or something else is going on ( nsp2 T85I, nsp13 P504L, spike S943P/T).
We are also working with @anekrut on parallel comprehensive analysis of intra-host variation from NGS data and linking the two levels of analysis (some of it is already there).
April 1st update.
SEVEN (maybe eight) sites currently showing some evidence of selection
- S D614G : has some reversions, toggle-like behavior; HLA/CTL?
- ORF3a Q57H : additional line of evidence – intrahost NGS variation data
- ORF8 84L/S : may be toggling
- ORF1a T265I (nsp2 T85I) : late and multiple introductions (high frequency)
- ORF1a L960F (nsp3 L142F) : late and multiple introductions (low frequency)
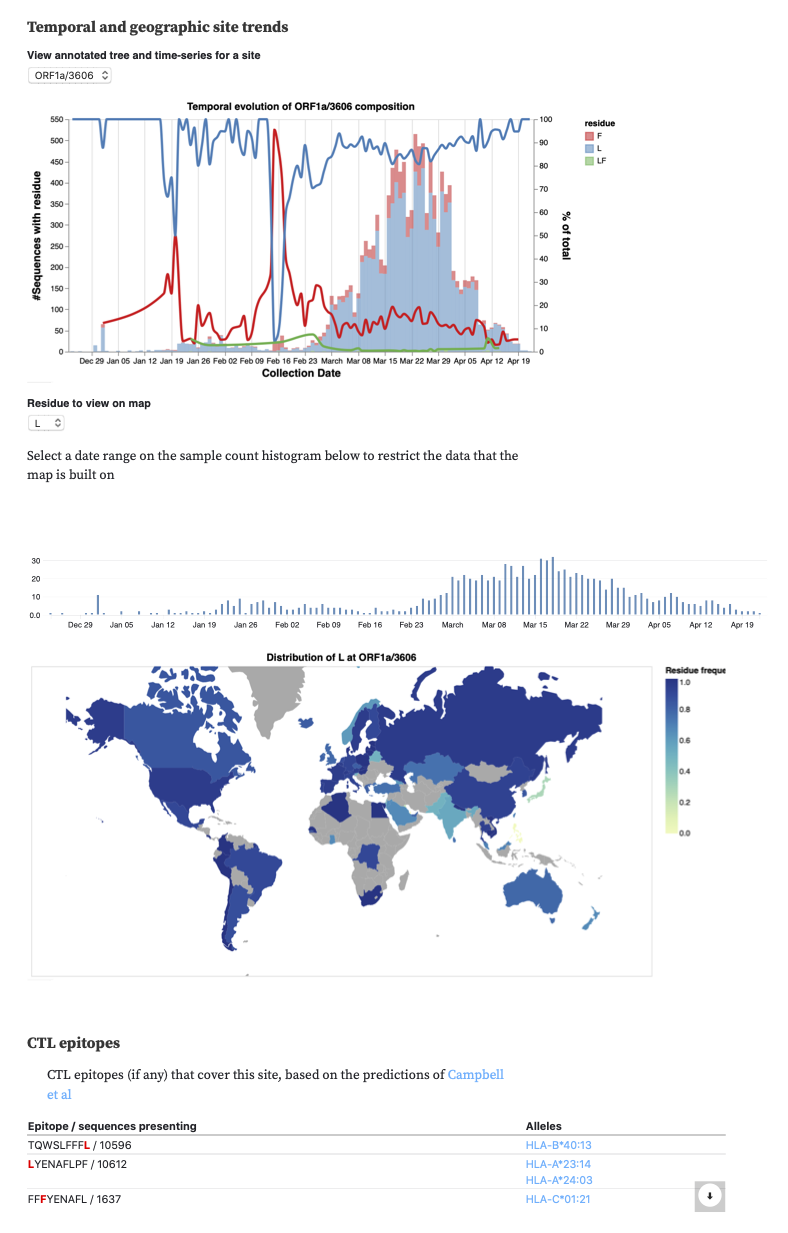
- ORF1a L3606F (nsp6 L37F) : additional line of evidence – intrahost NGS variation data
- ORF1b T1729I (exonuclease T206I) : late and multiple introductions (low frequency)
- (Maybe) ORF1a L1599F (nsp3 L781F) : this variant appears to have gone extinct
Details at http://covid19.datamonkey.org/2020/04/01/covid19-analysis/
1 Like
April 5th update.
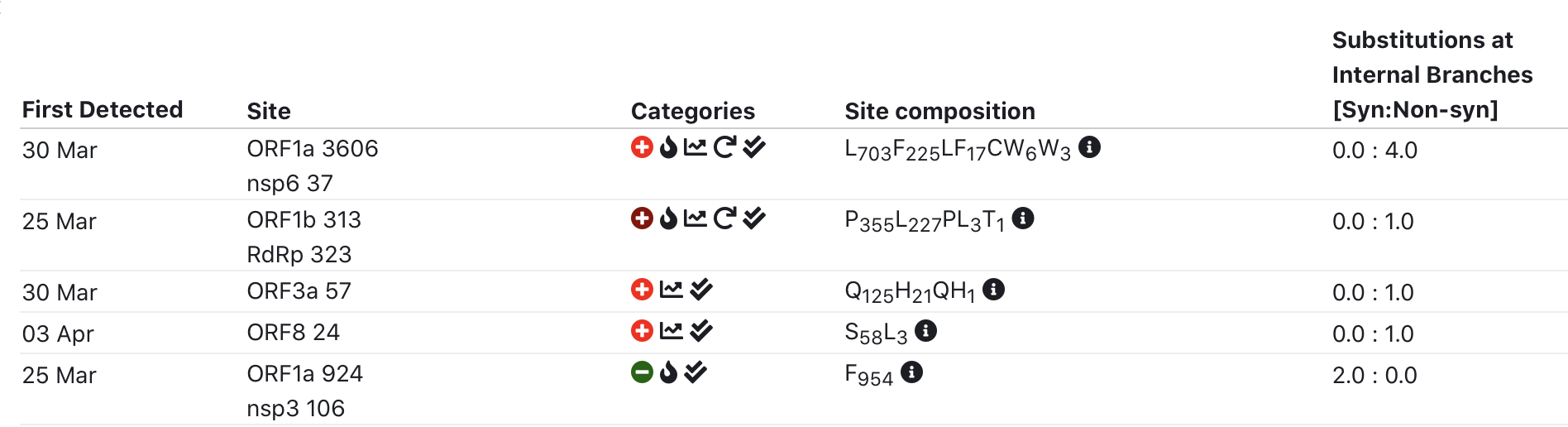
To facilitate interpretation, added a simple 5-category scoring system for sites, and some ways to summarize data in geographic format.
Update summary: https://twitter.com/sergeilkp/status/1247139846260432897
Details: http://covid19.datamonkey.org/2020/04/01/covid19-analysis/
Top 5 interesting sites:
May 4th update.
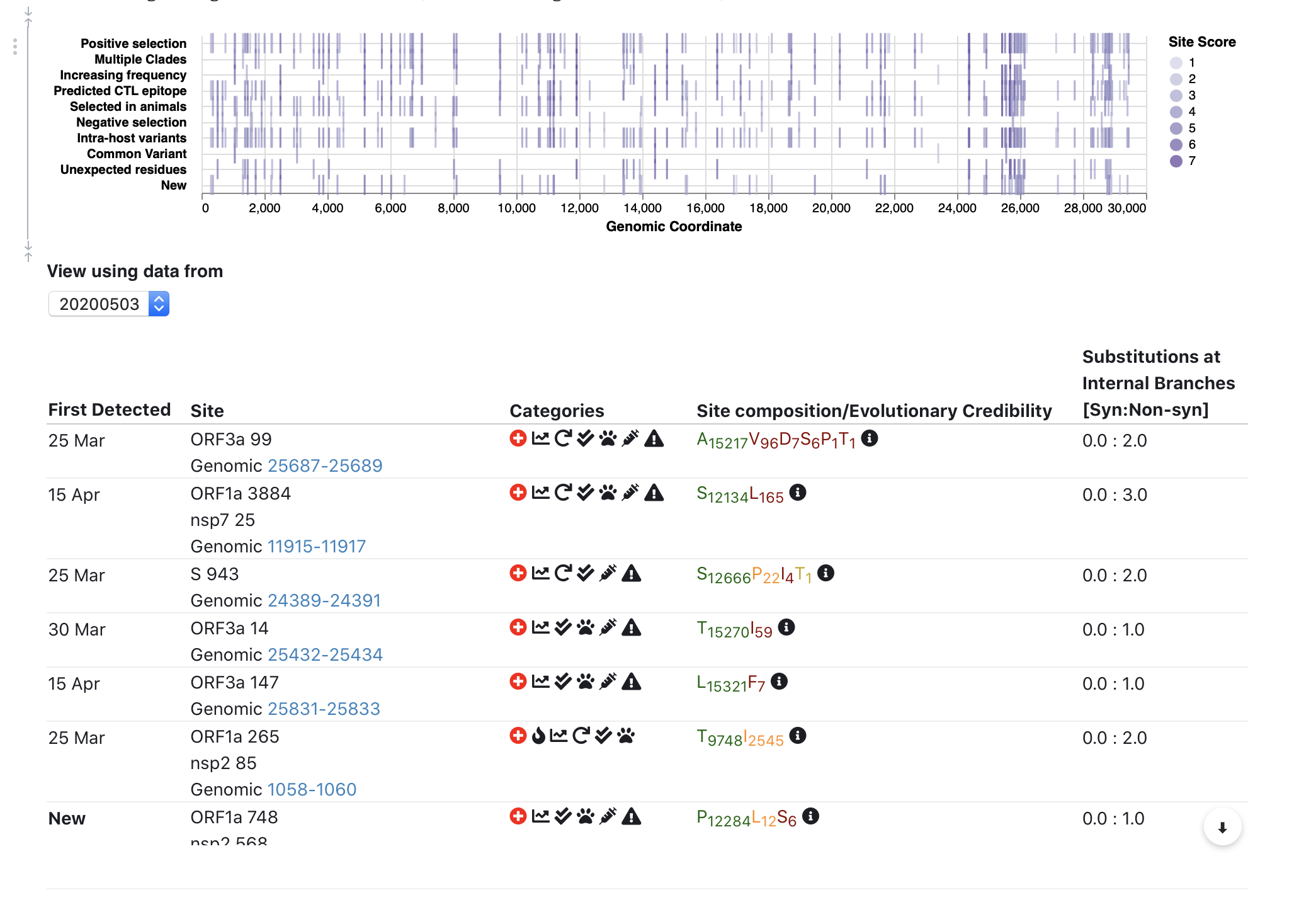
We expanded site annotation to include information about evolution in closely related beta-coronavirus lineages (bats, pangolins, etc); based on a collaborative effort with the @david.l.robertson @oscar.maclean and Spyros Lytras.
- Is the site selected in bat/pangolin beta-coronaviruses most closely related to SARS-CoV-2?
- Is the site evolving differently between closely and more distantly related bat strains?
We also incorporated data on CTL epitopes predicted using a comprehensive computational scan (Prediction of SARS-CoV-2 epitopes across 9360 HLA class I alleles | bioRxiv), so you can see which sites overlap which epitopes.
Our intra-host variation data now includes over 1000 SRA NGS data sets called using a standardized and publicly available pipeline (https://covid19.galaxyproject.org/genomics/4-Variation/).
Finally, if you are interested in incorporating our daily evolutionary annotation into your work, we provide a machine readable (JSON) file with site-level details at SARS-CoV-2/data at master · veg/SARS-CoV-2 · GitHub
1 Like