Recombinant SARS-CoV-2 genomes involving lineage B.1.1.7 in the UK
Ben Jackson1, Andrew Rambaut1, Oliver G Pybus2, David L Robertson3, Tom Connor4, Nicholas J Loman5, The COVID-19 Genomics UK (COG-UK) consortium6
1 University of Edinburgh
2 University of Oxford
3 MRC-University of Glasgow Centre for Virus Research
4 Cardiff University
5 University of Birmingham
6 https://www.cogconsortium.uk – full consortium authorship list is available here (492.7 KB)

This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
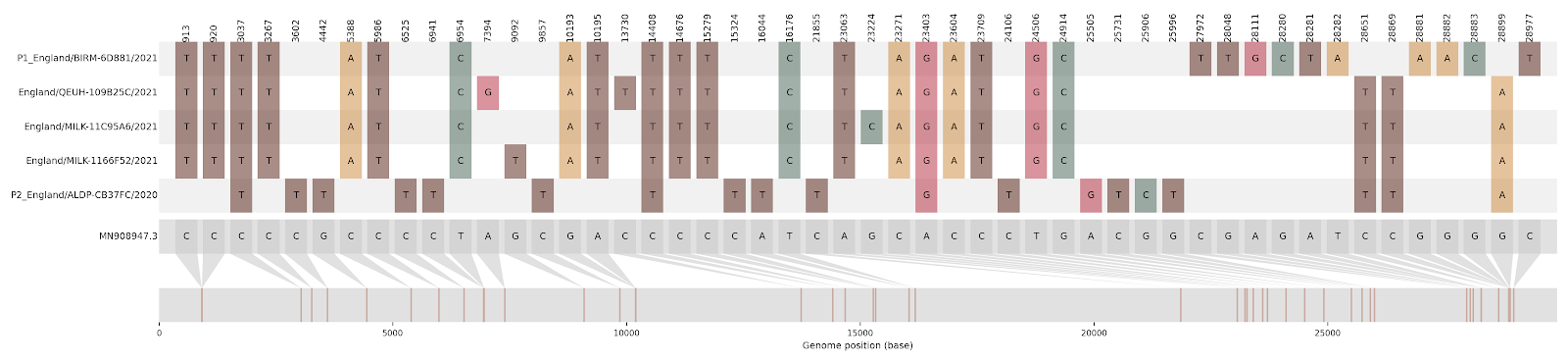
A survey of SARS-CoV-2 genome sequences from the UK has detected a number of variants that had been assigned to the B.1.1.7 lineage but which do not contain the full set of B.1.1.7 ‘lineage defining’ mutations [1]. Examination of these sequences revealed that some genome sections carry mutations characteristic of B.1.1.7, whilst other sections carry mutations specific to another lineage (Figure1; Table 1). Long runs of mutations along the SARS-CoV-2 that match different lineages are strongly indicative of virus recombination. In four instances (recombinant groups A-D) the same mosaic genome structure is observed in multiple closely-related genomes sampled from different infected people (Figure S1). These sequences are therefore highly unlikely to be artefactual (see discussion below). Additionally, we detected a further four mosaic virus genomes, each represented by only one genome sequence (Figure S2). These are also likely to be recombinants, but with a lower level of confidence.
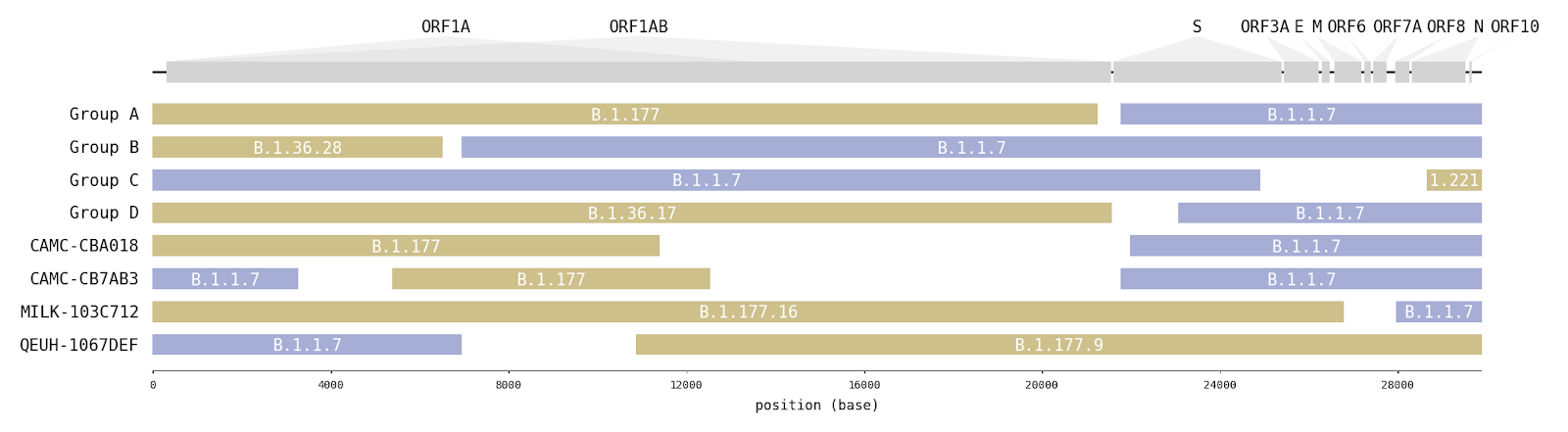
Figure 1 | Putative recombinant SARS-CoV-2 genomes and their inferred mosaic genome structures. Recombinant groups A-D contain multiple sequences exhibiting the same mosaic genome structures (see Table 1 for details). Tracts matching lineage B.1.1.7 are shown in blue, whilst virus genome regions matching other lineages are shown in yellow. Gaps represent ambiguity in the exact position of the recombinant breakpoints; these genome regions could have been inherited from either parental lineage (i.e. there are no lineage-defining mutations within these regions).
Interestingly, in 6 of 8 instances (and all 4 of the transmitting groups), the mosaic viruses contain a spike gene from the B.1.1.7 lineage and in four instances there is a proposed recombination breakpoint at or near the 5’ end of the spike gene (Figure 1).
| No. | Location | Sample dates | 2nd parent lineage | Break-point coordinates | Closest 2nd parent date | Closest 2nd parent location | |
|---|---|---|---|---|---|---|---|
| Group A | 3 | North Wales | 2021-01-30 - 2021-02-14 | B.1.177 | 21255 - 21764 | 2021-01-27 | North Wales |
| Group B | 2 | South East | 2020-12-23 - 2020-12-24 | B.1.36.28 | 6528 - 6954 | 2020-12-10 | Greater London |
| Group C | 3 | East Midlands | 2021-01-18 - 2021-01-30 | B.1.221.1 | 24914 - 28651 | 2021-01-02 | East Midlands |
| Group D | 3 | South East | 2021-02-02 - 2021-02-07 | B.1.36.17 | 21575 - 23063 | 2020-12-10 | South East |
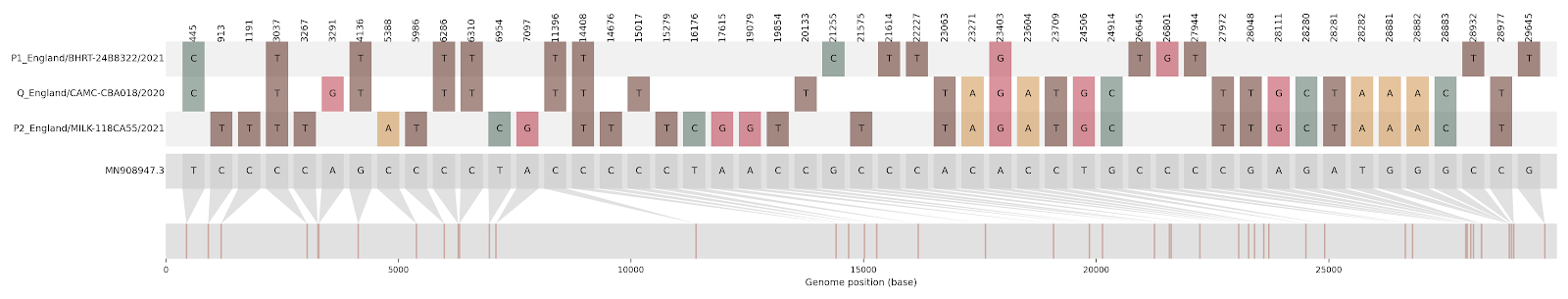
| CAMC-CBA018 | 1 | Greater London | 2020-12-18 | B.1.177 | 11396 - 21991 | 2021-01-23 | England |
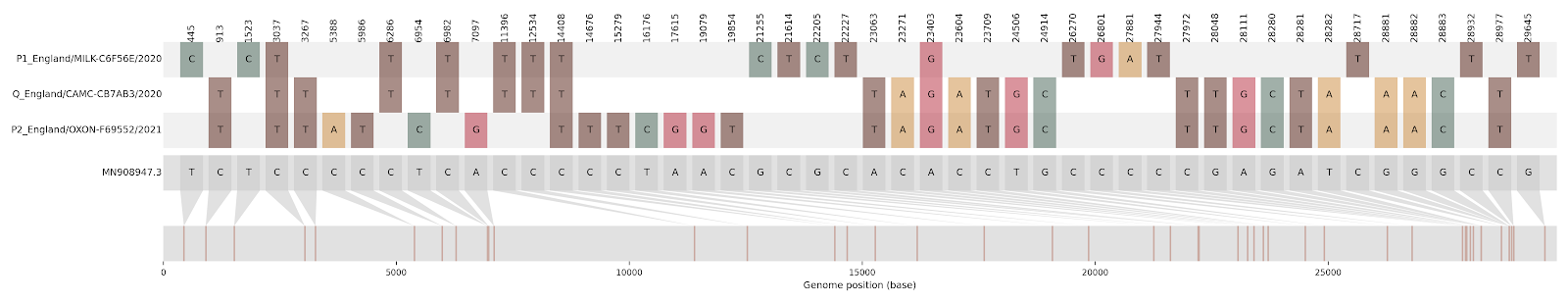
| CAMC-CB7AB3 | 1 | Greater London | 2020-12-18 | B.1.177 | 3267 - 5388, 12534 - 21765 | 2020-12-12 | Greater London |
| MILK-103C712 | 1 | Greater London | 2021-01-12 | B.1.177.16 | 26801 - 27972 | 2020-12-14 | Greater London |
| QEUH-1067DEF | 1 | Scotland | 2021-01-17 | B.1.177.9 | 6954 - 10870 | 2021-01-13 | England |
Table 1 | Recombinants and their putative second parent lineages (first parent lineage is B.1.1.7). For the recombinant groups the number of genomes, the NUTS1 location of residence and the range of sampling dates are given. Break-points are the range of possible nucleotide positions bounded by mutations that are unambiguously inherited from one parent or the other. The date and location for the 2nd parent is for the genetically most similar UK genome within the tract belonging to that lineage.
The timing, location, and genetic composition of the mosaic sequences detailed in Table 1 provides further, supporting evidence that they indeed originated through recombination in co-infected individuals in the UK. The generation of a recombinant SARS-CoV-2 genome requires an individual – and the same cell within that individual – to be co-infected with two genetically-distinct lineages, thereby allowing RNA template-switching to occur. Therefore recombination events are more likely to occur when prevalence is high. Additionally, the most likely parental lineages of a recombinant are those that are circulating at an appreciable frequency in the location where the recombinant was formed. Genomic surveillance of SARS-CoV-2 in the UK is widespread and representative, hence transmitted recombinant genomes can be detected relatively soon after they are generated.
The recombinants described here were likely generated within the UK during a period of high SARS-CoV-2 positivity, which was ~1%-2% of the general population in England between Oct 2020 and Jan 2021 (ONS Infection Survey; [2]. During this time, the B.1.1.7 variant of concern grew rapidly in relative frequency and spread across the UK, replacing previously-existing UK lineages that were already at high incidences [3], the most common of which were B.1.177 and its descendants (Figure 2). In all cases we describe here, the genomes of the putative parental lineages (B.1.1.7 and another second lineage – see Table 1) were present in the local geographic areas at the same time as the recombinants (e.g. North Wales for Group A). Specifically, B.1.36.28 was sampled 15 times between December and January, eight of which were in Greater London. B.1.221.1 was sampled 13 times in January, and all samples with detailed geographic information were from the Midlands of England. B.1.36.17 is represented by more than 100 samples from England in 2021, most of which lack more detailed geographic information.
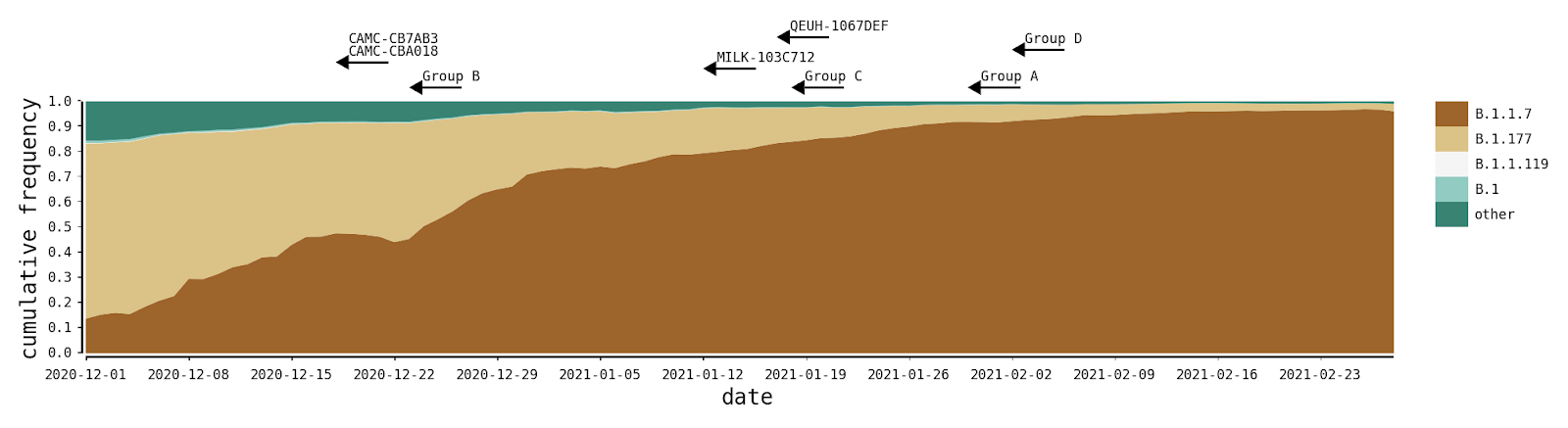
Figure 2 | The distribution of the most frequent SARS-CoV-2 lineages in the United Kingdom from December to February. Here, B.1.177 refers to B.1.177 itself and all its descendent lineages (e.g. B.1.177.9). For each recombinant, or recombinant group, the date of the earliest sampled genome is indicated by the left tip of the arrows. The recombination event that generated each must have occurred before this date.
Consideration of Alternative Explanations
The genetic distinctiveness of lineage B.1.1.7 (comprising 22 mutations across the genome, relative to the ancestral B.1.1 lineage; [1]) facilitated the identification of the recombinants reported here. Recombination between more genetically similar lineages would be difficult to distinguish with certainty. That said, it is worth exploring the range of plausible explanations for the mosaic genomes reported here.
One possible reason a genome sequence could have a mosaic structure is because it is a consensus of sequencing reads from different variants, present either because of laboratory contamination or because a mixture of virus lineages were present in the sample or patient. Whilst this can give rise to mosaic genomes, the sequencing protocol used in the UK [4] generates 98 short (~350bp) amplicons, such that long tracts that match just one lineage would be unlikely. For the groups A-D this possibility can be also excluded because multiple genomes with the same mosaic structure are sequenced independently from different samples, implying transmission of the recombinant has occurred. It should also be noted that, for group A, one of the three putative recombinant genomes was sequenced at a different facility than the other two.
For the putative recombinants with just a single representative it is possible that the sampled case is a co-infection and thus the sample is a mixture. As noted above, the short amplicon sequencing protocol used means that a coinfected sample is unlikely to generate a simple mosaic genome structure. Given that we observed four instances of transmitted recombinant genomes, we have no reason to suspect the four singletons are not also recombinants.
Discussion
Recombination is a frequent and well documented feature of the molecular evolution of coronaviruses [5], in the Sarbecovirus subgenus of SARS-CoV-2 [6–8] and has been inferred in SARS-CoV-2 specifically [9]. Recombination likely occurs in every infected individual, but in almost all cases recombination will be between nearly identical genomes (descendents of the small pool of viruses that established the infection).
Whilst the detection of clear examples of recombinant SARS-CoV-2 genomes in the UK is a consequence of the distinctness of the lineages involved and of the intensive genomic surveillance in the country, the frequency of coinfection will be a function of incidence and lineage co-circulation in a region. Given that many other countries are experiencing incidence levels that are comparable to those in the UK during late 2020 – early 2021, we predict that recombination between distinct lineages will be commonly detected wherever intensive genome sequencing is undertaken. Recombination between genetically-similar variants will be equally common but more difficult to detect.
However, like all coronaviruses, the appearance of recombinant SARS-CoV-2 genomes is expected and will have no immediate implications for the trajectory of the pandemic. Recombination of the B.1.1.7 lineage – identified as a ‘Variant of Concern’ – is likewise unremarkable as it rapidly became the dominant lineage across the UK whilst incidence was peaking so was most likely to be a constituent of any co-infection.
In the same way that individual amino acid replacements in the SARS-CoV-2 genome are mostly the result of error-prone replication, and are not expected to have phenotypic effects, most recombination events are simply the result of the within-host processes that occur in every infection.
The recombinant genomes reported here have not been designated a Pango lineage name because they do not meet the minimum observation size criterion [10]. If a recombinant virus does meet the criteria then provisional rules for naming these lineages have been posted here: https://pando.tools/t/657.
References
-
Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. 18 Dec 2020 [cited 9 Mar 2021]. Available: https://pando.tools/t/563
-
Coronavirus (COVID-19) Infection Survey, UK - Office for National Statistics. Office for National Statistics; 12 Mar 2021 [cited 17 Mar 2021]. Available: Coronavirus (COVID-19) Infection Survey, UK - Office for National Statistics
-
Volz E, Mishra S, Chand M, Barrett JC, Johnson R, Geidelberg L, et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv. 2021; 2020.12.30.20249034.
-
Tyson JR, James P, Stoddart D, Sparks N, Wickenhagen A, Hall G, et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. Cold Spring Harbor Laboratory. 2020. p. 2020.09.04.283077. doi:10.1101/2020.09.04.283077
-
Graham RL, Baric RS. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J Virol. 2010;84: 3134–3146.
-
Boni MF, Lemey P, Jiang X, Lam TT-Y, Perry BW, Castoe TA, et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. 2020;5: 1408–1417.
-
Hon C-C, Lam T-Y, Shi Z-L, Drummond AJ, Yip C-W, Zeng F, et al. Evidence of the Recombinant Origin of a Bat Severe Acute Respiratory Syndrome (SARS)-Like Coronavirus and Its Implications on the Direct Ancestor of SARS Coronavirus. J Virol. 2008;82: 1819–1826.
-
Hu B, Zeng L-P, Yang X-L, Ge X-Y, Zhang W, Li B, et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017;13: e1006698.
-
VanInsberghe D, Neish AS, Lowen AC, Koelle K. Recombinant SARS-CoV-2 genomes are currently circulating at low levels. doi:10.1101/2020.08.05.238386
-
Rambaut A, Holmes EC, Hill V, O’Toole Á, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. doi:10.1101/2020.04.17.046086
Supplementary Figures
a)
b)
c)
d)
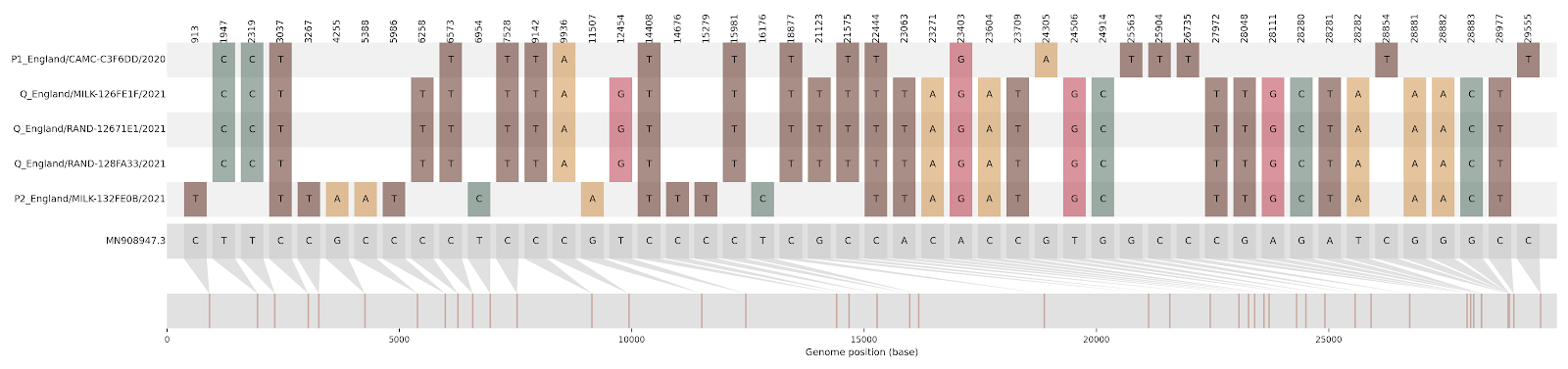
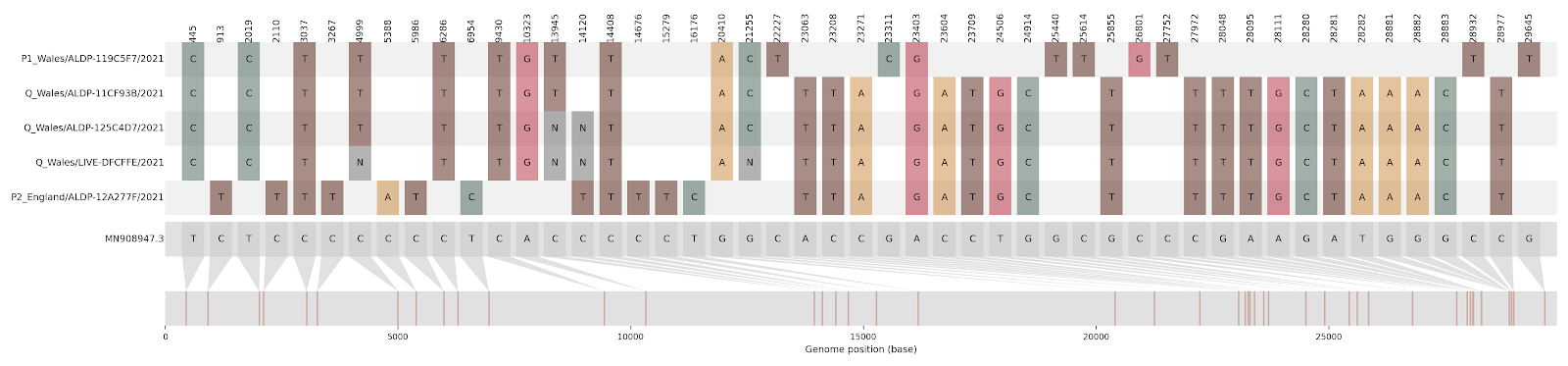
Figure S1 | The closest genome match to the first tract is shown at in at the top, and to the second tract at the bottom. Plot shows all nucleotide differences from the reference genome (accession MN90894). a) Group A – 3 genomes from Wales with similarity to lineage B.1.177 up until a break-point between nucleotide 21255 - 21764 and B.1.1.7 after this point. b) Group B – 2 genomes from South East England with similarity to lineage B.1.36.28 until between 6528 - 6954. c) Group C – 3 genomes from East Midlands with similarity to lineage B.1.221.1 after a break-point between 24914 - 28651. d) Group D – 3 genomes from South East England with similarity to lineage B.1.36.17 until 21575 - 23063. Figures generated using the snipet package by Áine O’Toole, GitHub - aineniamh/snipit: snipit: summarise snps relative to your reference sequence.
a)
b)
c)

d)

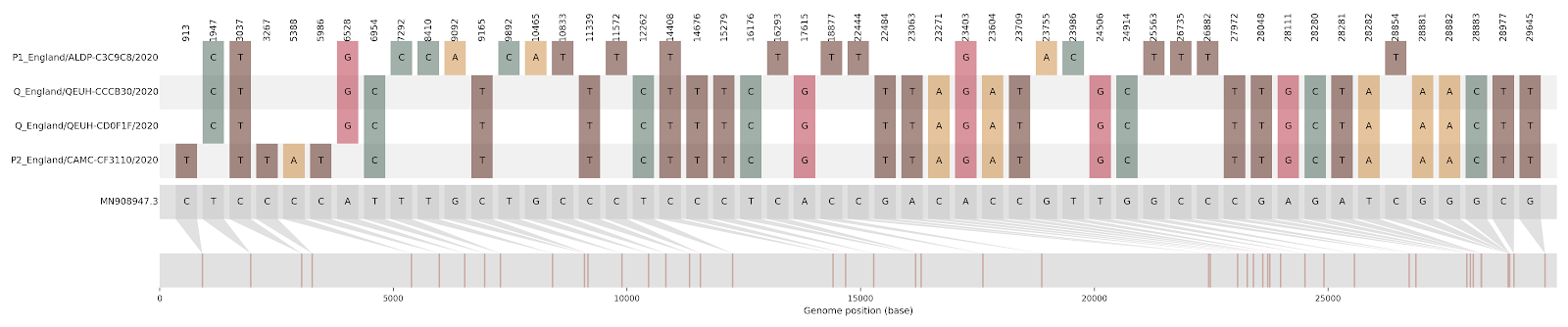
Figure S2 | Four putative recombinants represented by a single genome a) CAMC-CBA018 (second parent lineage B.1.177), b) CAMC-CB7AB3 (second parent lineage B.1.177) c) MILK-103C712 (second parent lineage B.1.177.16), d) QEUH-1067DEF (second parent lineage B.1.177.9)