Preliminary analysis of full genome sequences of 58 MPXV clade Ib cases from Kamituga and Kamanyola, South-Kivu , DRC
Recently, we showed that the MPXV outbreak in South Kivu is associated with a novel sub- lineage of clade I viruses (Murhula Masirika et al, 2024). The majority of these cases seem to be transmitted through heterosexual contact, with also person-to-person transmission within and outside the households. Here we provide an update of the ongoing sequencing effort. We sequenced MPXV genomes from samples during the current outbreak collected between October 2023 and May 2024. All samples were collected within a study protocol with ethics approval and informed consent in place.
58 complete, or nearly complete, whole genome sequences were generated. Phylogenetic analysis and inspection of the mutation patterns revealed three potential clusters and two potential subclusters consisting of sequences of different collection dates including April/May 2024, suggesting several different and ongoing transmission chains. Research efforts are ongoing to link the genomic data with epidemiological metadata (https://www.medrxiv.org/content/10.1101/2024.05.10.24307057v1).
During the course of this MPXV outbreak we observe an accumulation of APOBEC3 mutations indicative of human-to-human transmission. Most APOBEC3 mutations were observed in cluster A, that includes the two sequences from cases from Kamanyola, bordering Rwanda. These two cases are not directly linked to each other and show up to 10 unique mutations, suggestion considerable transmission prior to their detection.
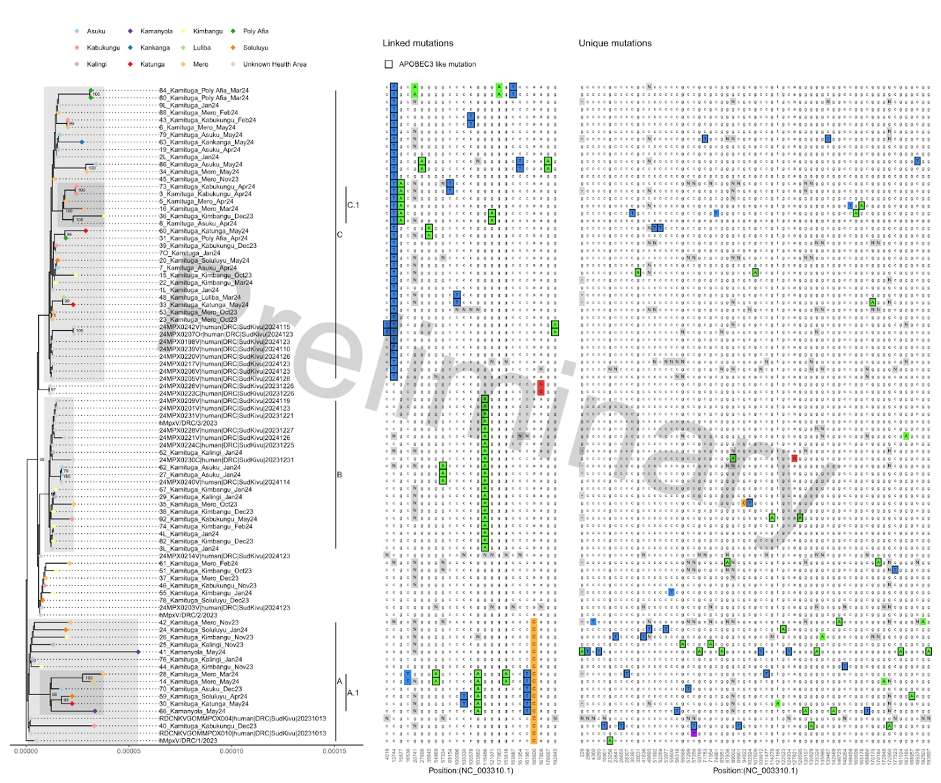
Figure 1: Zoom-in of phylogenetic tree specific for the currently shared and newly sequenced clade Ib sequences. Left panel shows the phylogenetic tree with colored tips indicating health area of originating patient. Proposed clusters are shown by shading and annotated by markings on the right side of the tree. Middle panel shows linking mutations, present in two or more cases. Right panel shows unique mutations. Differences from the majority rule consensus are highlighted in color. Mutations with characteristic APOBEC3 signature are marked with a black box. Positions that were masked by manual curation or due to too little coverage are indicated with “N”. Ambiguous nucleotides are marked in purple.
Contacts:
For more information on the ongoing studies, sequences and data analysis, please contact Leandre Murhula Masirika [email protected] , Bas Oude Munnink [email protected] and Marion Koopmans [email protected]
Authors and Funding:
Sequencing and data analysis were made possible by funding provided by the GREAT-LIFE consortium and South-Kivu government public health team supporting data collection.
The following individuals are involved in this work:
DRC: Leandre Murhula Masirika, Justin Bengehya Mbiribindi, Freddy Belesi Siangoli
Rwanda : Pacifique Ndishimye, Jean Claude Udahemuka, Jean Pierre Musabyimana
Denmark: Frank M. Aarestrup
Netherlands: David F. Nieuwenhuijse, Leonard Schuele, Marjan Boter, Bas B. Oude Munnink, Marion Koopmans
United Kingdom: Trudie Lang