Hi,
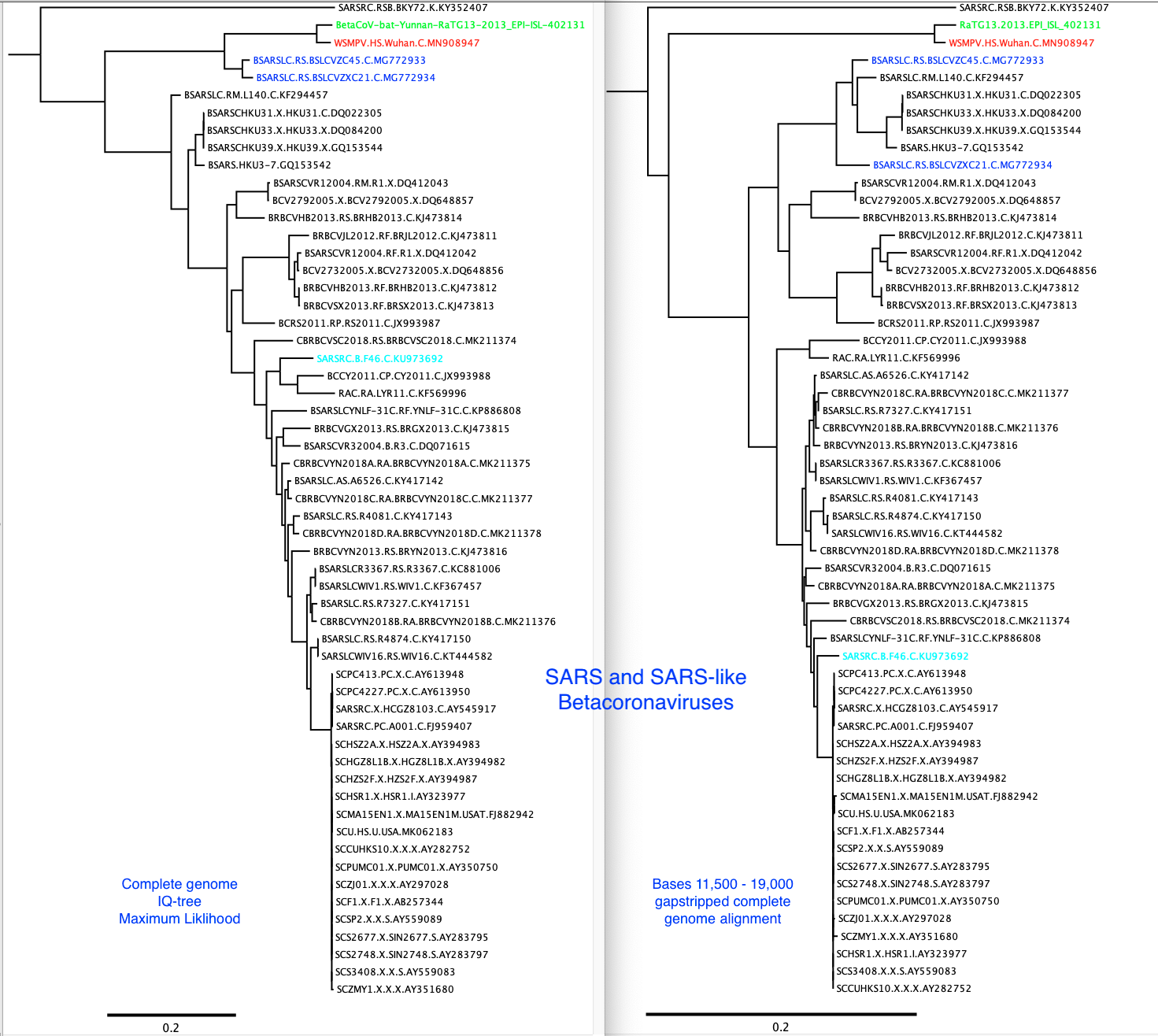
I have added the 2013 Yunnan bat sequence (much more closely related to the Wuhan2019-2020 virus than are the XC21 and ZC45 viruses) and built trees from complete genomes and some subgenomic regions chosen based on similarity plots.
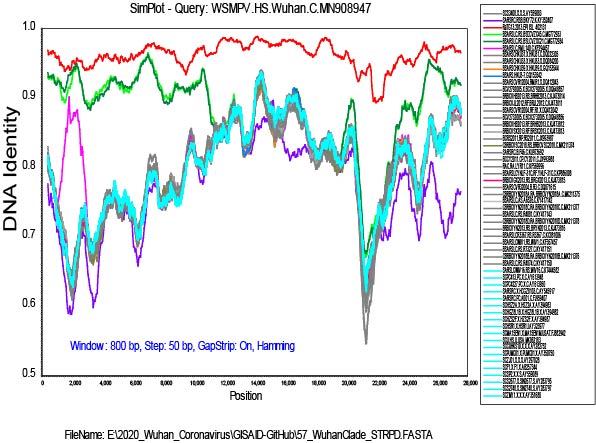
From this type of data, we cannot say which viruses are “recombinant” and which are “pure”
or never recombined in their history. It is only clear that recombination has been happening in the evolution of these viruses.
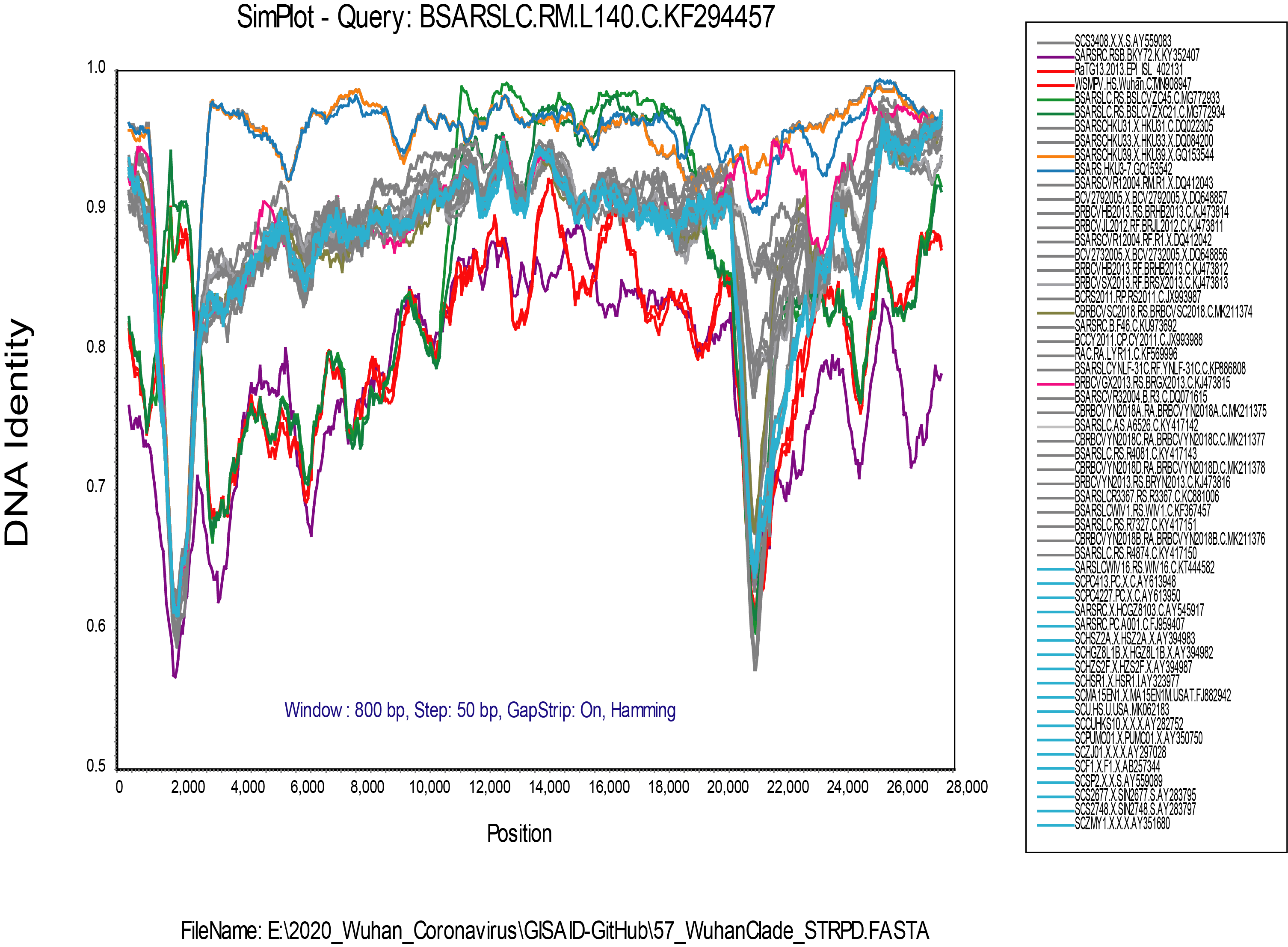
My alignment is built from a sampling of complete genomes of only the SARS and “SARS-like” subclade of the Betacoronaviruses. It does not include MERS or other Betacoronaviruses. For the SimPlot (Stuart Ray software) I first gap-stripped the alignment.
I am now running an RDP4 (Darren Martin software) alignment on the gap-stripped set, and it is finding a lot of recombination. The recombination events that I found evident in SimPlot are also detected in RDP4, but I am a new user of RDP4 and it will take me a while to get up to speed with using it.
Brian Foley
HIV Databases at LANL
1 Like
Just to be clear our first post on Jan 22nd was to highlight there’s no evidence the new human lineage nCoV was recombinant, rather the, at that time, known to be related bat coronaviruses are clearly the recombinants. Since then a pre-print has come out from Wuhan-based researchers with senior author Zheng-Li Shi: https://www.biorxiv.org/content/10.1101/2020.01.22.914952v1 including a similarity plot. In this very important study Zhou et al. report a closer bat coronavirus to the nCoV lineage, RaTG13, in all of its genome confirming the nCoV lineage is not recombinant. The bat coronaviruses are themselves recorded to be frequently recombinant in the published literature so while interesting this is not a new finding. Update, Zhou et al’s paper is now available in Nature: https://www.nature.com/articles/s41586-020-2012-7.
2 Likes
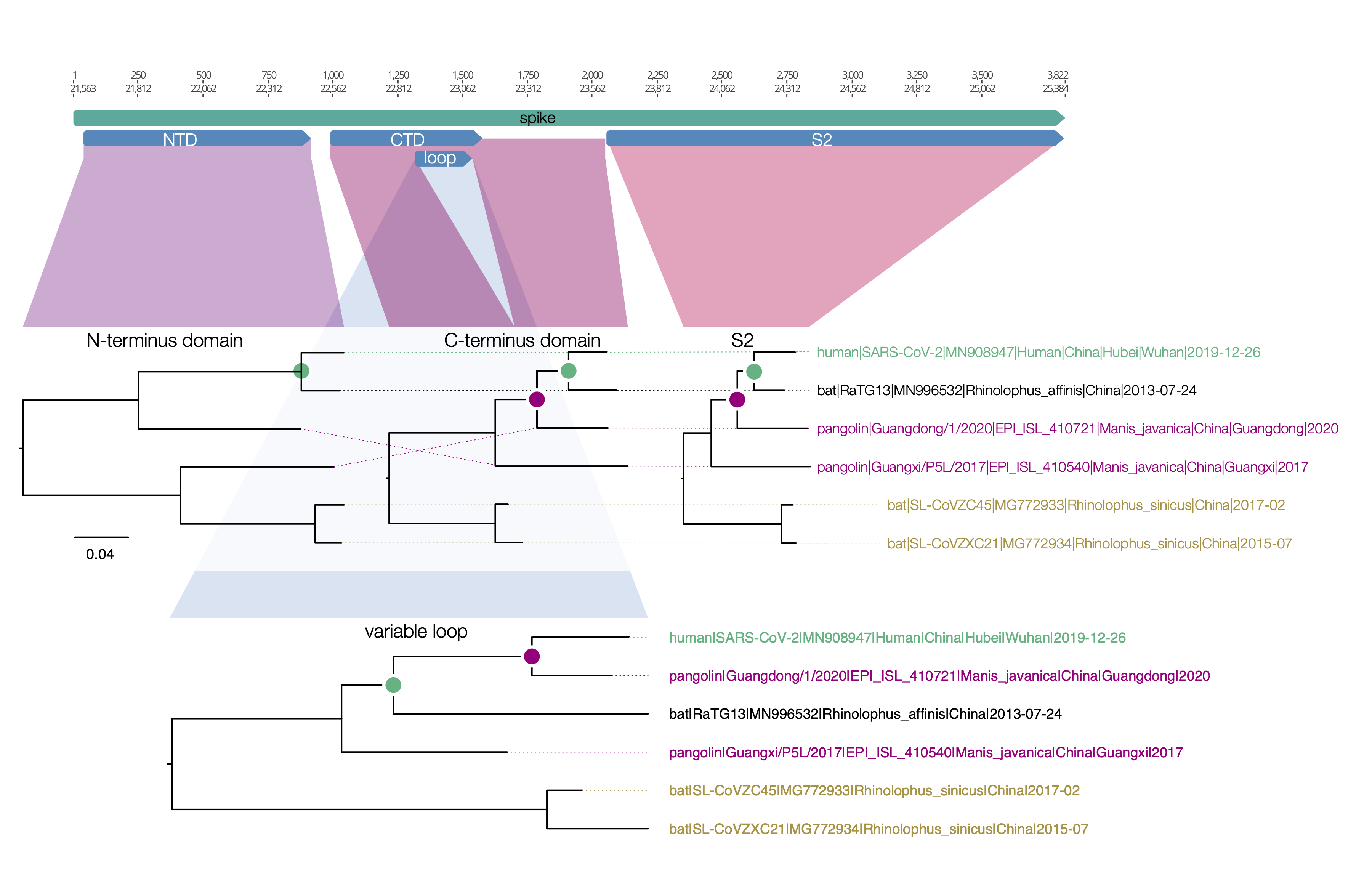
I have put together a figure of the recombination pattern of some of the closest viruses to SARS-CoV-2 including RaTG13 and the pangolins. Includes the region indexed as 1680-3014 by David, above, although I call the breakpoints as 1455-2836. The entire region after the spike is just lumped together.
Figure 1 | Phylogenetic trees for regions across the genome of SARS-CoV-2 and related betacoronaviruses.
spike protein
Here is a similar diagram for the spike protein. Generally the bat virus, RaTG13, is the closest to the SARS-CoV-2 virus across the whole spike with the exception of the small variable loop region in the C-terminus domain (the receptor binding domain). In this region, RaTG13 suddenly leaps away in divergence leaving the pangolin virus, Guangdong/1/2020 as the closest. This suggests that RaTG13 acquired this different loop region by recombination with another bat virus.
Figure 1 | Regions of the spike protein SARS-CoV-2 and its closest relatives. Trees are drawn to the same scale.
What is interesting about this loop region is that it contains the six key contact residues for the ACE2 receptor (see this post for more details about the RBD). So this suggests the common ancestor of the RaTG13 virus, the pangolin and the SARS-CoV-2 had the optimal receptor binding domain for ACE2 and then RaTG13 lost it.
2 Likes
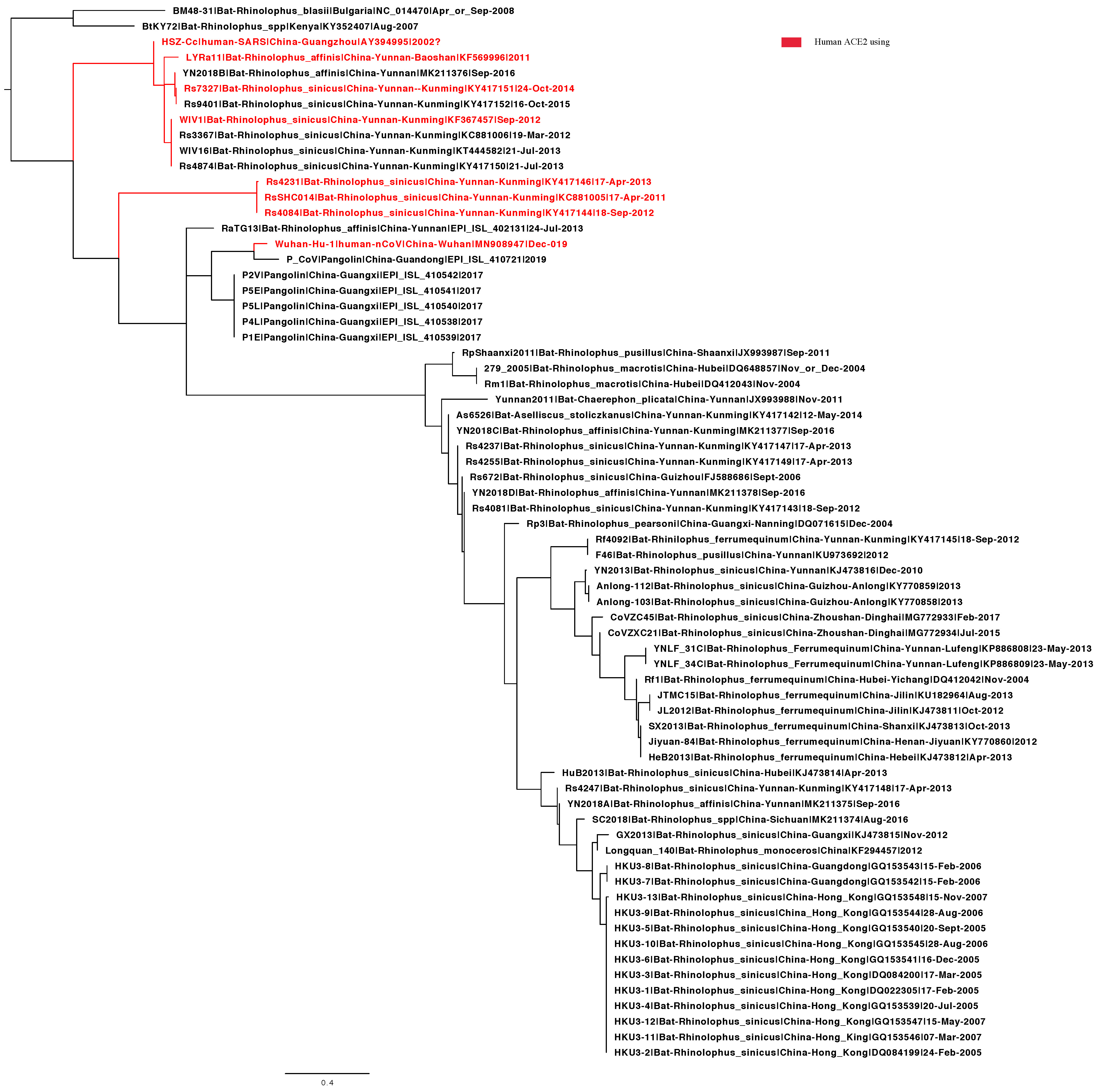
The phylogenetic tree of this loop is indeed interesting (ACE2 binding motif). We have been working on this too. Using the same data set as we used above with six new pangolin sequences, we find that the clustering with ACE2 using bat strains (labelled red, experimentally verified see Functional assessment of cell entry and receptor usage for lineage B β- coronaviruses, including 2019-nCoV) suggests the ability to use human ACE2 may have been pre-adapted before jumping to humans in a bat species or there may be other regions close to RBD involved to determine ACE2 usage.
The pangolin strain, which causes clinical symptoms in pangolin, may come from a bat species originally. We may still need to find the actual intermediate animal.
1 Like