Data here.
Protocol here.
Blog post here.
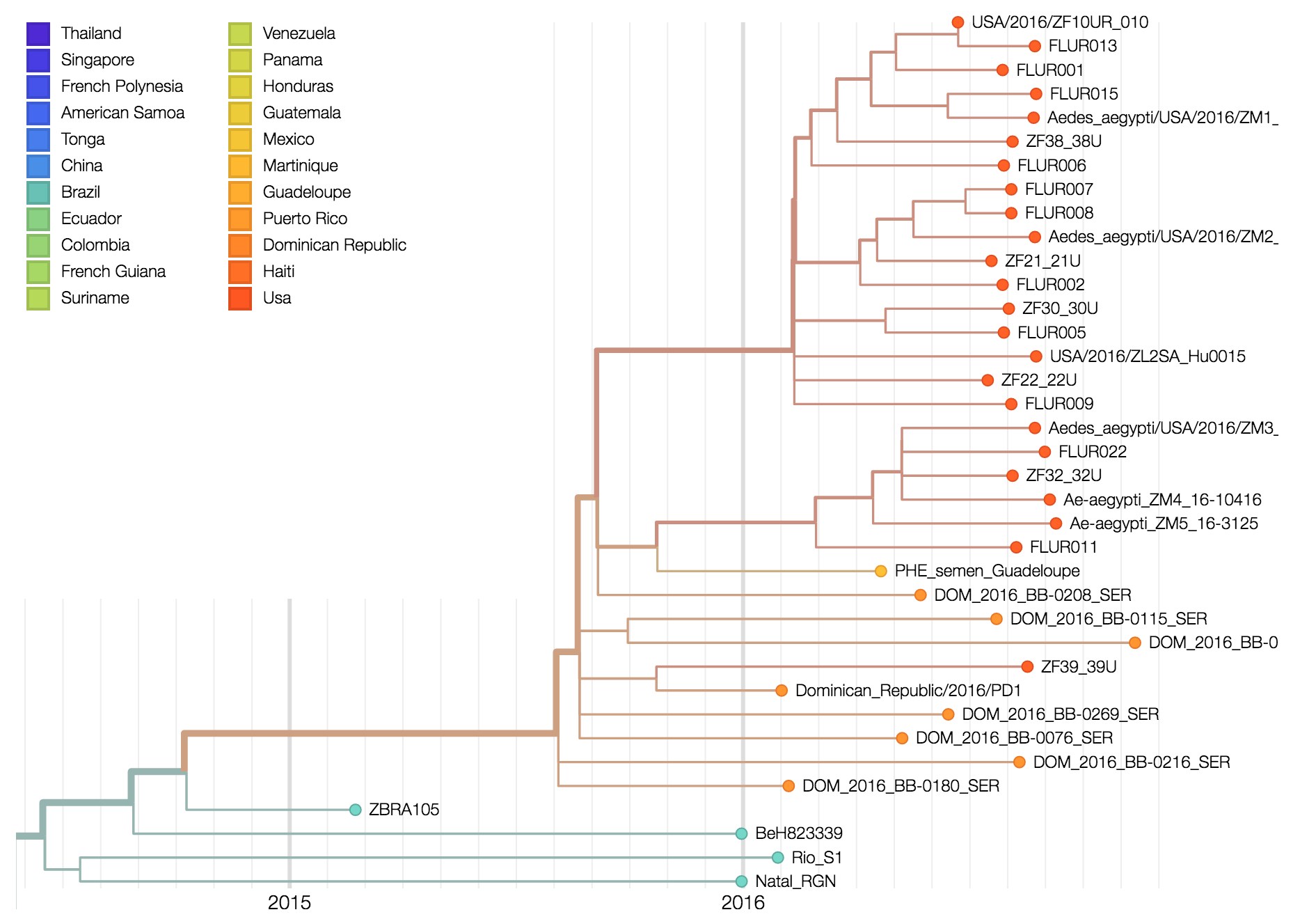
In collaboration with Diogo Magnani from the University of Miami, we (the Andersen Lab) recently received plasma and saliva from two people with Zika virus infections living in the Miami area. Using our amplicon-based approach previously used to sequence Zika virus from travel-related Zika virus cases in Florida, we sequenced the entire coding sequence from the saliva of one individual believed to have been infected in or near Miami. This sequence (ZL2_Hu0015) is related to other Zika viruses recently sequenced from the Americas (Figure 1), indicating that the current Zika virus outbreak in Florida was the result of an imported infection (human or mosquito) from the ongoing epidemic in the Americas. It is not a result of a separate introduction from Africa or earlier circulating Asian strains. ZL2_Hu0015 is 99.95% identical (5 mismatches) to a Zika virus that we recently sequenced from a traveler coming into Florida from Cuba (ZF10_010U) on June 2nd, 2016. We did not detect any contaminating reads from ZF10_010U in our ZL2_Hu0015 data set (libraries prepared separately with different reagents), demonstrating that these are independent samplings of related viruses. While the data is suggestive, we do not have enough data at this time to conclusively determine the direct origin of the virus. Further sequencing of Zika viruses recovered from autochthonous transmission will help to resolve if there were more than one Zika virus introductions contributing to the outbreak in Florida.
We did, however, detect some contamination in our sequencing reads belonging to PRVABC59, our commonly used lab control. While we are working to remove contamination, we believe that the contamination did not alter the consensus sequences we are reporting here.
In collaboration with Scott Michael and Sharon Isern from Florida Gulf Coast University, we also recently sequenced ZIKV from travel-associated cases in Florida, including the first sequence from Cuba. Data here. Blog post here.
Update #1 (11Sep16)
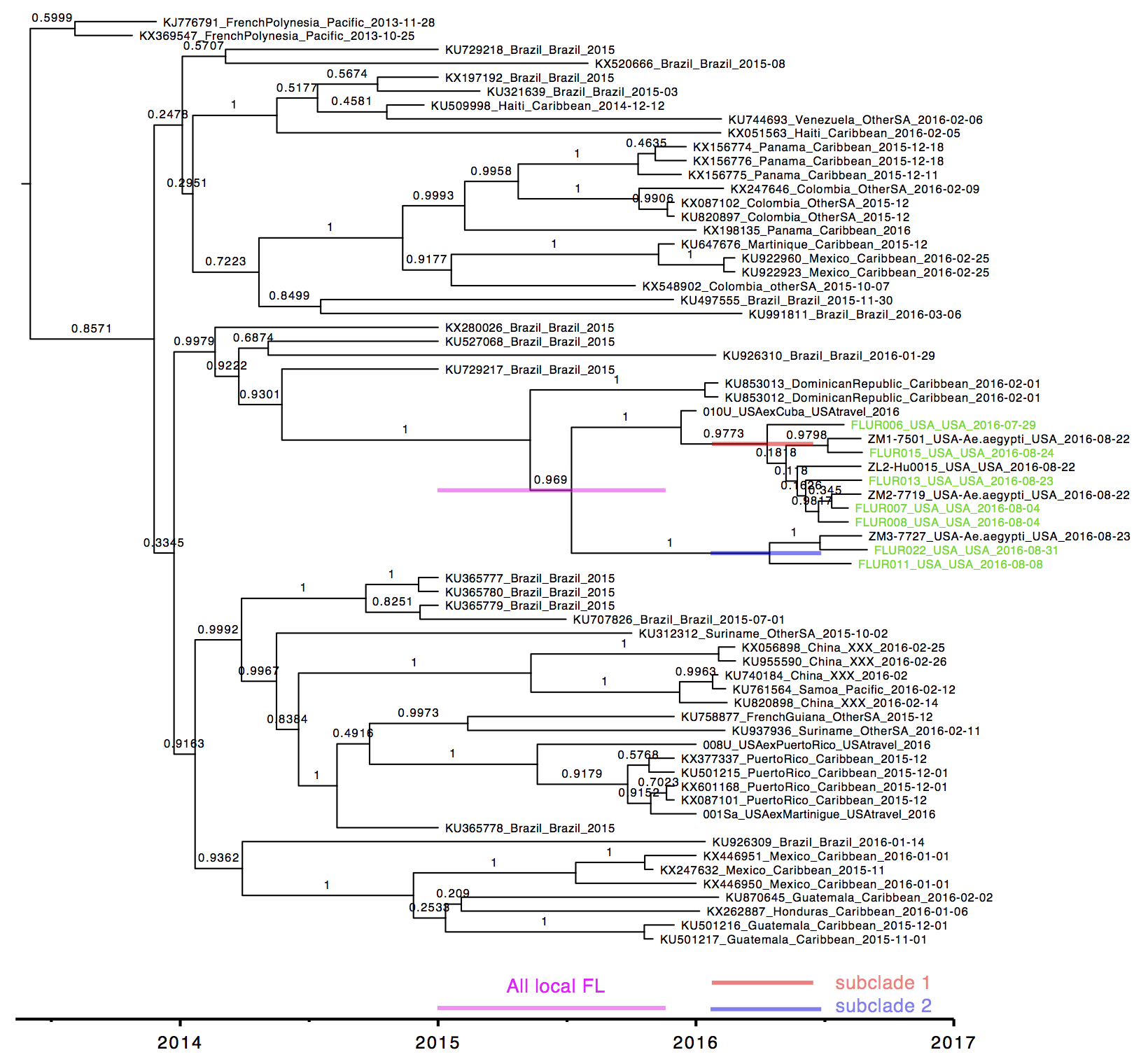
Three pools of Aedes aegypti mosquitoes collected in Miami Beach on August 22nd and 23rd, 2016, were found to be infected with Zika virus. Through our collaborators, Scott Michael and Sharon Isern from Florida Gulf Coast University, we recently received samples of these mosquitoes for sequencing using our amplicon-based protocol for MiSeq. We obtained ~7.5 million 250 bp Zika virus reads per sample, giving us a mean genome coverage of ~160,000 nucleotides per site. Previous cross-contamination issues have been mostly resolved as we only detected 346 reads aligning to Zika virus in our water controls. The Zika virus genomes obtained from the mosquitoes formed a distinct clade (strong bootstrap support) with a Zika virus sequenced from a Miami infection and a traveler returning to Miami from Cuba. The close relationship between the sequences suggests that 1) there is Ae. aegypti-borne Zika virus transmission in the Miami area, 2) the traveler from Cuba may have been actually infected in Miami, and 3) the outbreak was initiated by a single Zika virus introduction from the ongoing epidemic in the Americas. At this point, however, it is difficult to determine the exact origin of the introduced virus. More Zika virus data are needed to help resolve the tree.

Zika virus tree was created using RAxML. Blue = Zika virus-infected Ae. aegypti collected in Miami Beach, red = suspected local human infection, and purple = suspected travel-associated infection.
We will be sequencing additional clinical samples from Florida Zika virus infections this week. Data will be released ASAP.
Feedback encouraged and openly welcome.
Nate