Summary
A complete genome sequence was obtained for a severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) isolated from a South African patient with coronavirus disease 2019 (COVID-19), who had returned to South Africa after traveling to Italy.
Genome Report
COVID-19, a disease caused by SARS-CoV-2 (Zhou et al., 2020) is spreading rapidly in South Africa (South African Department of Health and the NHI, 2020), the rest of the African continent (Africa CDC, 2020) and the world (World Health Organization, 2020). Next-generation sequencing of pathogens can provide new insights into disease transmission and aid in drug and vaccine design (Gwinn et al., 2019). Since 2008, the Global Initiative on Sharing All Influenza Data (GISAID) has emerged as a platform for international sharing of influenza virus sequence data, and more recently SARS-CoV-2 data (Shu and McCauley, 2017). As of 01 April 2020, more than 3000 SARS-Cov-2 genomes were globally sequenced and uploaded to GISAID (The GISAID Initiative, 2020). The Nextstrain (The Nextstrain Team, 2020) website provides real-time monitoring of the spread and evolution of the SARS-CoV-2 virus, drawing on sequence data stored in GISAID. As a contribution to the global efforts to track and trace the ongoing coronavirus pandemic, here we present the sequence, phylogenetic analysis and modelling of non-synonymous mutations for a SARS-CoV-2 genome that was detected in a South African patient with COVID-19.
Nasopharyngeal and oropharyngeal swabs from a symptomatic individual were collected and combined. Total nucleic acid extraction was performed using the MagNA Pure 96 DNA and Viral NA Small Volume Kit (Roche, Switzerland) as described by the manufacturer. SARS-CoV-2 nucleic acid was detected using the TIB Molbiol LightMix Sarbeco E-gene real-time polymerase chain reaction assay, which yielded a cycle threshold (Ct) value of 23.21 (Corman et al., 2012) A subsequent nucleic acid extraction, obtained using the QIAamp Viral RNA Mini Kit (QIAGEN, Germany), was assessed for total RNA quantity and integrity with the Qubit RNA Assay Kit (Invitrogen, Carlsbad, CA, USA) and Agilent 4200 TapeStation (Agilent Technologies, Germany). Host Ribosomal RNA depletion was performed using NEBNext rRNA depletion kit (New England Biolabs, Ipswich, MA, USA), followed by cDNA synthesis. The paired-end libraries were prepared using the Nextera DNA Flex library preparation kit, followed by 2×300 bp sequencing on Illumina MiSeq (Illumina, San Diego, CA, USA). All above mentioned methods and techniques were done at the National Institute for Communicable Diseases, a division of the National Health Laboratory Service, Johannesburg, South Africa.
The resultant metagenomic sequence reads (9,406,678 paired-end reads) were quality trimmed (Q>20) using Trim Galore (Felix Krueger, 2019) and subsequently FastQ Screen (Steven Wingett, 2019) was used to filter out non-viral reads. The remaining viral reads (23,489 reads) were then mapped to the complete genome of SARS-CoV-2 Wuhan-Hu-1 isolate (Genbank accession number: MN908947.3) using CLC Bio (Qiagen, 2020) to generate the consensus sequence. The consensus sequence was combined with a collection of 965 SARS-CoV-2 genomes downloaded from GISAID and a multiple sequence alignment (MSA) was generated using MAFFT v7.042 (Katoh and Standley, 2013) running within Nextstrain (Hadfield et al., 2018) at the South African National Bioinformatics Institute (SANBI), University of the Western Cape, Cape Town, South Africa. The sequence reads used to generate the consensus were mapped against the MN908947.3 sequence using BWA-MEM v0.7.17 (Li, 2013) running in Galaxy (Afgan et al., 2018).
Variants in the consensus sequence were identified by inspecting the MSA, validated by inspecting the read mapping and visualised in IGV (Robinson et al., 2011). From an initial list of 74 variants, 6 were confirmed by the evidence from mapped reads and retained. Depth of coverage was computed using samtools (Li et al., 2009), and averaged over the genome, yielding an average depth of 10 reads. Regions of high (greater than 5 reads) coverage were identified using covtobed (Birolo and Telatin, 2020) and the resultant BED file used to mask variant positions (using Python and the intervaltree module in a Jupyter notebook (Thomas et al., 2016)). This masking confirmed that the previously mentioned 6 good quality variants were located within the 76% of the genome that was covered by reads to a depth greater than 5. Variants were inserted into the MN908947.3 reference sequence using BioPython (Cock et al., 2009). The variants identified were as follows (with position in genome, nucleotide change, number of reads supporting/number of reads mapped, gene name, amino acid change):
241 C → T 15/16 in 5’ UTR
3037 C → T 13/13 in orf1ab / nsp3 (AA 193: synonymous Phe)
13620 C → T 6/6 in orf1ab / nsp12 (AA 58: synonymous Asp)
14408 C → T 18/18 in orf1ab / nsp12 (AA 321: Pro → Lue)
21595 C → T 7/7 in S (spike) protein (AA 10: synonymous Val)
23403 A-> G 6/6 (spike) protein (AA 614: Asp → Gly)
The variants at 13620 and 21595 were not found in any other SARS-CoV-2 genome that was present in GISAID at the time that this report was drafted.

Figure 1: SARS-CoV-2 Spike (S) trimer modelled with SWISS-MODEL (Waterhouse et al., 2018), drawn and coloured in PyMol (DeLano, W. L. (2009). Domains of a single S1 protomer are shown in cartoon view and coloured green (N-terminal domain, NTD), red (C-terminal domain/receptor binding domain, CTD/RBD), purple (subdomain 1 and 2, SD1 and SD2). S2 is shown in dark teal, while N-acetylglucosamine moieties are coloured yellow (cartoon protomer) or orange (surface protomers). A zoomed inset shows the location of D614, which is where a mutation has arisen in the R03006/20 South African strain, buried in the interprotomer interface.
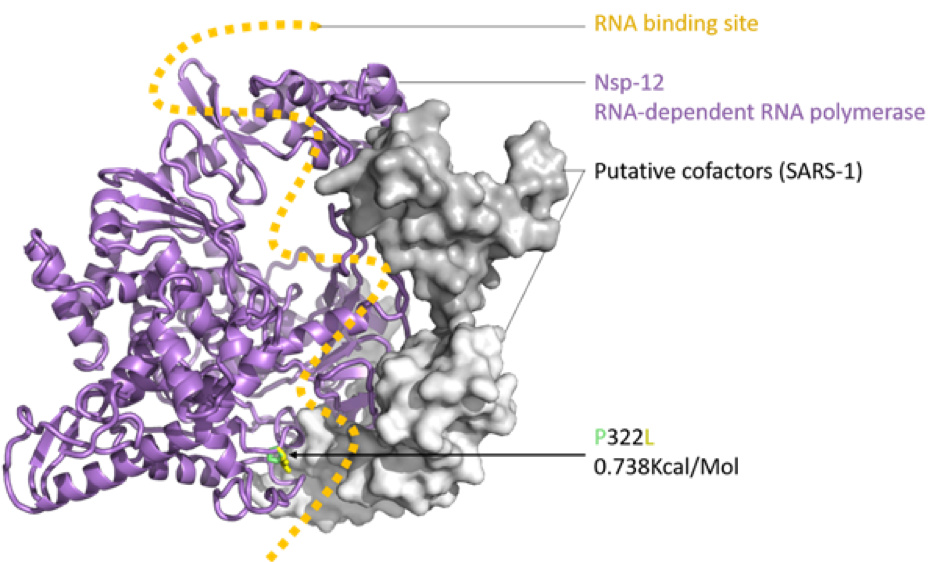
Figure 2: SARS-CoV-2 nsp12 (RNA-dependent RNA polymerase, RDRP) modelled with SWISS-MODEL (Waterhouse et al., 2018), drawn and coloured in PyMol (DeLano, W. L. 2009), based on the nsp12, nsp7 and nsp8 protein complex of SARS-1 (PDB: 6NUR). The RNA binding groove is indicated (orange), with adjacent P332 (green) to L332 (yellow) mutation shown in stick view.
The impact of the S protein D614G variant (Figure 1) and the P322L variant on the nsp12 protein (Figure 2) were predicted by the DUET webserver (Pires et al. 2014) to have slightly destabilising and stabilising effects, respectively.
Neither the receptor binding domain (RBD) of the S protein nor the points of contact between the nsp12 and the putative SARS-CoV-2 co-factors nsp7 and nsp8 were impacted by these variants, leading to the assumption that overall the variants won’t have a substantial effect on protein structure or function. Figures were produced with PyMol (maintained by Schrödinger, LLC., 2019).
Analysis by Nextstrain showed that the genome clustered together with other genomes from Europe and the United States of America, supporting the evidence that the epicenter of the COVID-19 pandemic is now found in these regions.
Contributing authors
Mushal Allam1, Arshad Ismail1, Zamantungwa T. H. Khumalo1, Stanford Kwenda1, Peter van Heusden2, Ruben Cloete2, Constantinos Kurt Wibmer1, Thabo Mohale1, Kathleen Subramoney1, Sibongile Walaza1, Wendy Ngubane1, Nevashan Govender1,Nkengafac V. Motaze, Jinal N. Bhiman1, on behalf of the SA-COVID-19 response team
Affiliations
1 National Institute for Communicable Diseases of the National Health Laboratory Service, Johannesburg, South Africa, 2131
2 South African Medical Research Council Bioinformatics Capacity Development Unit, South African National Bioinformatics Institute, University of the Western Cape, Cape Town, South Africa. 7735
Acknowledgments
We thank the SA-COVID-19 response team at the National Institute for Communicable Diseases of the National Health Laboratory Service, South Africa and the National Department of Health, South Africa. Special thanks goes to Dr Sandile Tshabalala the head of the KwaZulu-Natal Department of Health and his team for facilitating sample collection. Work at SANBI-UWC was supported by the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation of South Africa (64751 to Alan Christoffels) and the South African Medical Research Council.
References
Afgan, E., Baker, D., Batut, B., van den Beek, M., Bouvier, D., Čech, M., Chilton, J., Clements, D., Coraor, N., Grüning, B.A., Guerler, A., Hillman-Jackson, J., Hiltemann, S., Jalili, V., Rasche, H., Soranzo, N., Goecks, J., Taylor, J., Nekrutenko, A., Blankenberg, D., 2018. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update | Nucleic Acids Research | Oxford Academic
Africa CDC, 2020. COVID-19 Daily Updates. Afr. CDC. URL Coronavirus Disease 2019 (COVID-19) – Africa CDC (accessed 3.27.20).
Birolo, G., Telatin, A., 2020. covtobed: a simple and fast tool to extract coverage tracks from BAM files. J. Open Source Softw. 5, 2119. https://doi.org/10.21105/joss.02119
Cock, P.J.A., Antao, T., Chang, J.T., Chapman, B.A., Cox, C.J., Dalke, A., Friedberg, I., Hamelryck, T., Kauff, F., Wilczynski, B., Hoon, M.J.L.D., 2009. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 25, 1422–1423. Biopython: freely available Python tools for computational molecular biology and bioinformatics | Bioinformatics | Oxford Academic
Corman, V.M., Eckerle, I., Bleicker, T., Zaki, A., Landt, O., Eschbach-Bludau, M., van Boheemen, S., Gopal, R., Ballhause, M., Bestebroer, T.M., Muth, D., Müller, M.A., Drexler, J.F., Zambon, M., Osterhaus, A.D., Fouchier, R.M., Drosten, C., 2012. Detection of a novel human coronavirus by real-time reverse-transcription polymerase chain reaction. Eurosurveillance 17. https://doi.org/10.2807/ese.17.39.20285-en
Felix Krueger, 2019. Babraham Bioinformatics - Trim Galore! [WWW Document]. URL Babraham Bioinformatics - Trim Galore! (accessed 3.25.20).
Gwinn, M., MacCannell, D., Armstrong, G.L., 2019. Next-Generation Sequencing of Infectious Pathogens. JAMA 321, 893. https://doi.org/10.1001/jama.2018.21669
Hadfield, J., Megill, C., Bell, S.M., Huddleston, J., Potter, B., Callender, C., Sagulenko, P., Bedford, T., Neher, R.A., 2018. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123. Nextstrain: real-time tracking of pathogen evolution | Bioinformatics | Oxford Academic
Katoh, K., Standley, D.M., 2013. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability | Molecular Biology and Evolution | Oxford Academic
Li, H., 2018. bwa: Burrow-Wheeler Aligner for short-read alignment (see minimap2 for long-read alignment).
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R., 1000 Genome Project Data Processing Subgroup, 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. Sequence Alignment/Map format and SAMtools | Bioinformatics | Oxford Academic
Pires, D.E.V., Ascher, D.B., Blundell, T.L., 2014. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 42, W314–W319. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach | Nucleic Acids Research | Oxford Academic
Qiagen, 2020. CLC Genomics Workbench [WWW Document]. Bioinforma. Softw. Serv. QIAGEN Digit. Insights. URL https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-clc-genomics-workbench/ (accessed 4.1.20).
Robinson, J.T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E.S., Getz, G., Mesirov, J.P., 2011. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. Integrative genomics viewer | Nature Biotechnology
Schrödinger, LLC., 2019. The PyMOL Molecular Graphics System, Version 2.3.
Shu, Y., McCauley, J., 2017. GISAID: Global initiative on sharing all influenza data – from vision to reality. Eurosurveillance 22, 30494. https://doi.org/10.2807/1560-7917.ES.2017.22.13.30494
South African Department of Health and the NHI, 2020. COVID-19 South African coronavirus news and information portal [WWW Document]. SA Corona Virus Online Portal. URL https://sacoronavirus.co.za/ (accessed 3.27.20).
Steven Wingett, 2019. Babraham Bioinformatics - FastQ Screen [WWW Document]. URL Babraham Bioinformatics - FastQ Screen (accessed 3.25.20).
The GISAID Initiative, 2020. GISAID - Global Initiative on Sharing All Influenza Data [WWW Document]. URL https://www.gisaid.org/ (accessed 3.27.20).
The Nextstrain Team, 2020. Nextstrain [WWW Document]. URL auspice (accessed 3.27.20).
Thomas, K., Benjamin, R.-K., Fernando, P., Brian, G., Matthias, B., Jonathan, F., Kyle, K., Jessica, H., Jason, G., Sylvain, C., Paul, I., Damián, A., Safia, A., Carol, W., Team, J.D., 2016. Jupyter Notebooks - a publishing format for reproducible computational workflows. Stand Alone 87–90. IOS Press Ebooks - Jupyter Notebooks – a publishing format for reproducible computational workflows
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F.T., de Beer, T.A.P., Rempfer, C., Bordoli, L., Lepore, R., Schwede, T., 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. SWISS-MODEL: homology modelling of protein structures and complexes | Nucleic Acids Research | Oxford Academic
World Health Organization, 2020. Coronavirus disease 2019 (COVID-19)Situation Report –66.
Zhou, P., Yang, X.-L., Wang, X.-G., Hu, B., Zhang, L., Zhang, W., Si, H.-R., Zhu, Y., Li, B., Huang, C.-L., Chen, H.-D., Chen, J., Luo, Y., Guo, H., Jiang, R.-D., Liu, M.-Q., Chen, Y., Shen, X.-R., Wang, X., Zheng, X.-S., Zhao, K., Chen, Q.-J., Deng, F., Liu, L.-L., Yan, B., Zhan, F.-X., Wang, Y.-Y., Xiao, G.-F., Shi, Z.-L., 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273. A pneumonia outbreak associated with a new coronavirus of probable bat origin | Nature