It has been speculated that several factor may limit our ability to accurate estimate the evolutionary rate of ZIKV including: potential recombinant sequences, sequence integrity and multiple passage history. Two previous studies also showed that significantly higher rates of nucleotide substitution appeared to occur during urban rather than during enzootic transmission of arboviruses (Sall AA, Faye O, Diallo M, Firth C, Kitchen A, Holmes EC (2010) Yellow fever virus exhibits slower evolutionary dynamics than dengue virus. J Virol 84: 765-772. Volk SM, Chen R, Tsetsarkin KA, Adams AP, Garcia TI, Sall AA, et al. (2010) Genome-scale phylogenetic analyses of chikungunya virus reveal independent emergences of recent epidemics and various evolutionary rates. J Virol 84: 6497-6504).
We have estimated the nucleotide substitution rate of ZIKV E gene by Bayesian MCMC analyses using four different datasets: 1) the ZIKVCOMPLETE that included all ZIKV sequences availables at the time of this study; 2) the ZIKVNONREC that included all ZIKV sequences selected minus those previously characterized as recombinant in the original Faye et al. (2014) publication 3) the ZIKVHUMAN that only included non-recombinant ZIKV sequences retrieved from human; and 4) the ZIKVNON-HUMAN that only included non-recombinant ZIKV sequences retrieved from mosquitos and monkeys.
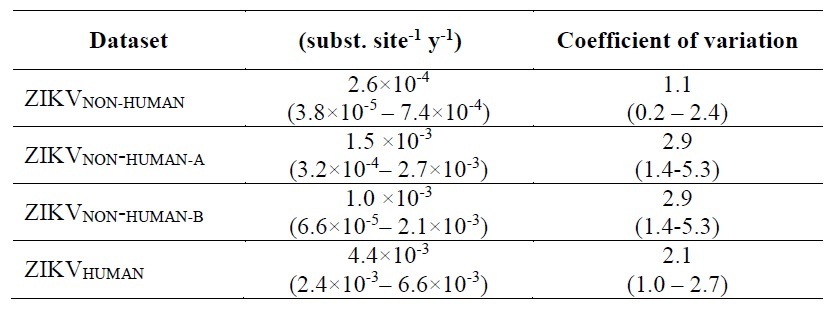
We found that the median substitution rates estimated from ZIKVCOMPLETE and ZIKVNONREC were nearly equal (1.0×10-3 susbs./site/year) (Figure), indicating that ZIKV molecular clock estimates were not affected by the inclusion of putative recombinants strains. The median evolutionary rate estimated for the ZIKVHUMAN data set (4.4×10-3 susbs./site/year), however, was more than 10 times higher than that estimated from ZIKVNON-HUMAN (2.6×10-4 susbs./site/year), whereas intermediate rates were obtained for those datasets combining ZIKV sequences from both human and non-human hosts (ZIKVCOMPLETE and ZIKVNONREC) (Figure).
These results supports the notion that significantly higher rates of nucleotide substitution appeared to occur during urban rather than during enzootic transmission of arboviruses and further indicates that the time-scale of ZIKV outbreaks in human populations should be reconstructed using only sequences from urban epidemics.
Using the ZIKVHUMAN data set, we estimated the TMRCA of ZIKV outbreak epidemic in the Americas in 2014 (95% HPD: 2013-2015) (see post http://virological.org/t/phylogeographic-analyses-point-to-a-single-introduction-event-responsible-for-the-2015-zika-virus-outbreak-in-the-americas/211). This estimate is nearly equal to that described by Nuno Faria using a dataset of contemporaneous NS5B gene sequences that was mostly composed (91%) by ZIKV sequences from humans (see post http://virological.org/t/zika-virus-introduction-in-the-americas-when-and-from-where/204). Thus, accurate estimates of the time-scale process of recent ZIKV dissemination in human populations could be obtained from both E and NS5 genes, since datasets were exclusively or mostly composed by viral sequences sampled from urban epidemics.
A manuscript is under preparation by several authors including Daiana Mir 1, Gonzalo Bello 1, Matthew T. Aliota 2 and Edson Delatorre 1 1. Laboratório de AIDS & Imunologia Molecular, Instituto Oswaldo Cruz, FIOCRUZ, Rio de Janeiro, Brazil. 2. Department of Pathobiological Sciences, School of Veterinary Medicine, University of Wisconsin-Madison.
This rate (4.4E-3) seems high to me. Also the ZIKVnon-human looks like it is simply recovering the prior (i.e., has no temporal information). I am going to put my money on the 1.0x10^-3 as the rate of ZIKV evolution in a sustained human-mosquito cycle (irrespective of whether it is urban or else where). But it would be useful if you could post which viruses you are using in each of these sets - it is difficult to make comparisons with the other estimates.
@edward_holmes - didn’t you mention the older sequences (the sentinel monkey and mosquito ones) had been passaged a lot?
The ZIKV-human dataset comprises sequences between 1968 and 2015. The ZIKV-Non-human dataset includes sequences from 1947 to 2002. We can maybe removed the older sequences with multiple passages from ZIKV-Non-human and estimate again the substitution rate. Which sequences from monkey and mosquito should be removed?
The evolutionary rate of the ZIKV-Non-human dataset (that mostly include sequences from the African lineages) was estimated at 2.6×10-4 subst/site/year (95% HPD: 3.8×10-5 – 7.4×10-4), that is remarkably similar to those previously estimated for other arboviral lineages mostly maintained in enzootic transmission cycles like YFV (2.1x10 4 substitutions/site/year, 95% HPD, 1.0-3.3x10 4) (Sall et al, 2010), and the CHIKV lineages WAf (2.4x10-4 subs/nt/year; 95% HPD: 2.0-2.8x10-4 subs/nt/year) and ECSA lineages (2.3x10-4 subs/nt/year; 95% HPD: 1.4-3.2x10-4 subs/nt/year).
On the other hand, the substitution rate estimated from the ZIKV-Human dataset that mostly include sequences of the Asian lineages (4.4×10-3 subst/site/year, 95% HPD: 2.4–6.6×10-3) was, indeed, much higher than those estimated from arboviral epidemic lineages like DENV (7.0-8.7x10 4 substitutions/site/year, range of 95% HPD, 5.8-9.8 x 10 4) (Sall et al, 2010), and the CHIKV lineages Asian (4.2x10-4 subs/nt/yr; 95% HPD: 3.3-5.0x10-4) and Indian Ocean (8.5x10-4 subs/nt/year; 95% HPD: 5.8-10.9x10-4 subs/nt/year).
This may indicate different selection pressures acting on the epidemic versus enzootic transmission cycles of ZIKV, and/or that the ZIKV-Human substitution rate was artificially increase because the intensive sampling of sequences from the recent Pacific and American ZIKV epidemics within a very short time span (2013-2015) may have included many transient deleterious mutations that would not persist over longer time periods.
A lognormal prior with mean 2.0 E-3 was applied on the clock rate of all the analyses on the basis of estimations reported from Faye et al. (2014). So the evolutionary rate estimated for the ZIKVNON-HUMAN dataset (2.6 E-4 susbs./site/year) did not recover the prior.
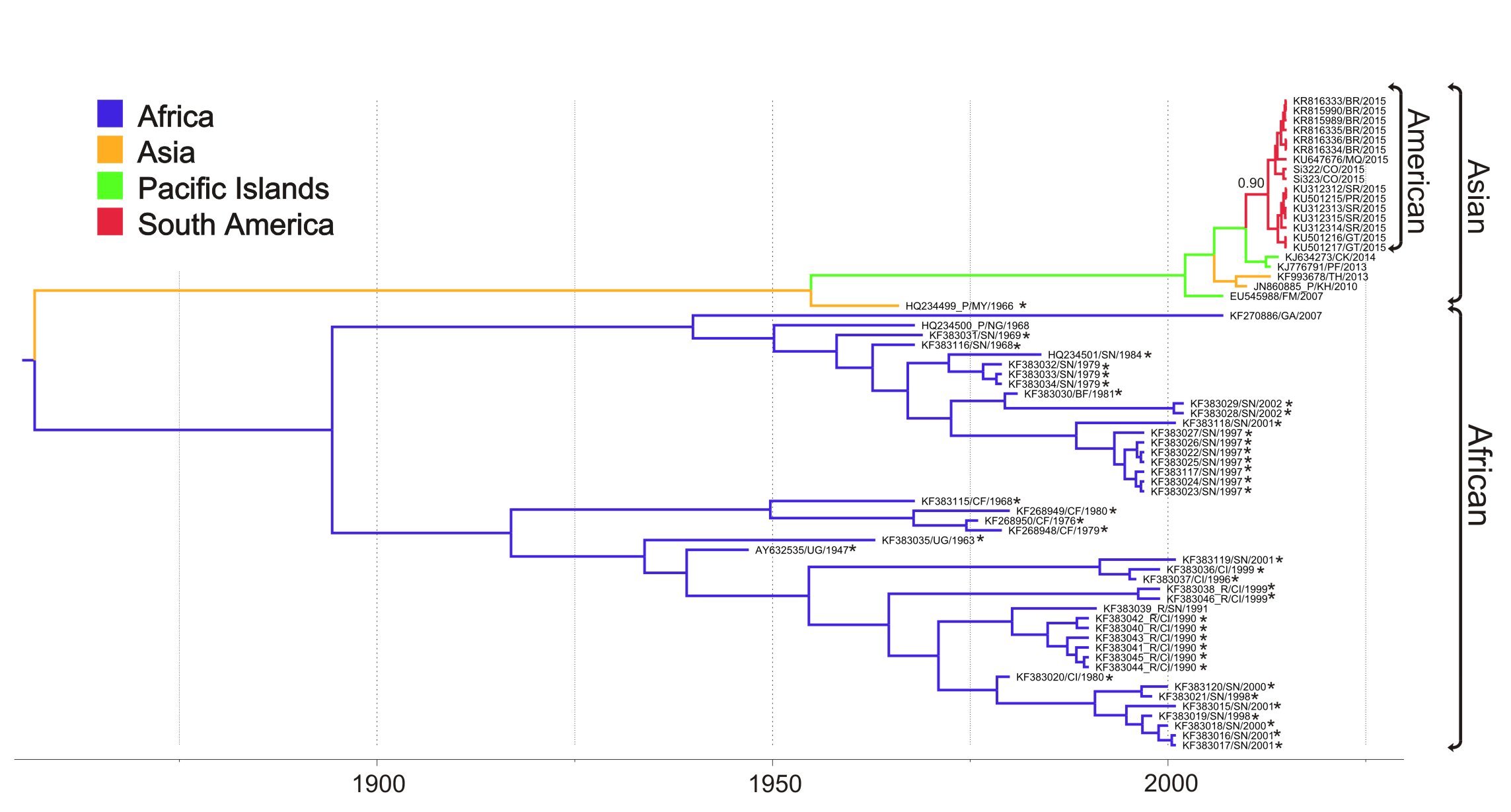
Below I place the figure of our MCC tree inferred for the ZIKVCOMPLETE dataset with non-human sequences highlighted with an asterisk. The accessions with _R highlight the ZIKV sequences characterized as recombinant in the original Faye et al. (2014) publication and those with _P highlight the ones with multiple passage history.
[quote=“daianamir, post:5, topic:212”]
So the evolutionary rate estimated for the ZIKVNON-HUMAN dataset (2.6 E-3 susbs./site/year) did not recover the prior.
[/quote] Do you mean 2.6 E-4 here?
Yes.
(1) Some of the older strains have been extensively passaged. I would be very cautious about using those.
(2) There are clearly issues with some of the sequences in the Faye et al. paper (that I have pointed out the authors). First, some of the recently sampled Senegalese strains are almost identical to one of the 1947 Uganda viruses. Second, there are a cluster of 6 viruses with a huge branch lengths and which greatly elevate the rate. Remove these.
(3) Also, you would expect high rates in recently sampled because of the presence of transient deleterious mutations; in other words, these rates often show a strong time-dependency. So, I think it is dangerous to conclude that rates are elevated in urban setting without account for time-dependency.
If I were to bet on the long rate in humans I would use DENV as a model and hence say somewhere between 0.5 - 1 x 10-3.
So the inferred rate was even lower than the prior mean rate? I really don’t think this rate is coming from the data. The interval spans more than an order of magnitude. I would suggest trying some different priors (try lognormal mean of 2E-4 and 2E-5) and checking the rate is robust to them. Also you could try the CTMC rate reference prior.
Thank you very much Rambaut and Holmes for your suggestions. We are going to test with different rate priors and sequences data. In the mean time, in your opinion, what dataset should be used to estimate the recent introduction of ZIKV in the Americas? One containing only recent human samples (with the potential risk to include transient deleterious mutations) or a dataset containing sequences from both human and non-human hosts (with the potential risk to be combining different viral ecological dynamics)?
I’d be tempted to wait for more sequences. You don’t want to have a repeat of the Ebola story and inane debates about what are in reality small differences in rate.
I have been looking at the full-length sequences on NCBI and it seems to me that ALL sequences prior to 2015 have been passaged in T/C. Here’s a rundown of the currently available genomes:
Year / country / # Passages (at least):
AY632535: 1947 / Uganda / 3x
EU545988: 2007 / Micronesia / Likely 0x, but chimera from four patients
HQ234501: 1984 / Senegal / 3x
JN860885: Cambodia / 2010 / 1x
KF268948: CAR / 1976 / ?x
KF383115: CAR / 1968 / ?x
KF383116: Senegal / 1968 / x?
KF383117: Senegal / 1997 / x?
KF383118: CAR / 2001 / ?x
KF383119: CAR / 2001 / ?x
HQ234499: Malaysia / 1966 / 740x
KF993678: Canada / 2013 / ?x
KJ776791: French Polynesia / 2013 / 3x
KU312312: Suriname / 2015 / Likely not passaged
KU321639: Brazil / 2015 / Likely not passaged
KU365777-80: Brazil / 2015 / Likely not passaged
There are a few MR766 strains (the one from ATCC) that have been passaged multiple times (3x - 747x - and they’re really quite different!) so I guess we could try and estimate the viral ‘clock rate’ in T/C and modify/calibrate the year-over-year substitution rates using that information?
I wouldn’t estimate T/C rate and use it to modify ‘natural’ rates. I think that would be asking for trouble. Some of these earlier strains are being re-sequenced I believe so I would wait for these. On the subject of Uganda a colleague in the know told me “The prototype Uganda strain has extensive passage in SM (>120) and likely is mouse adapted.”
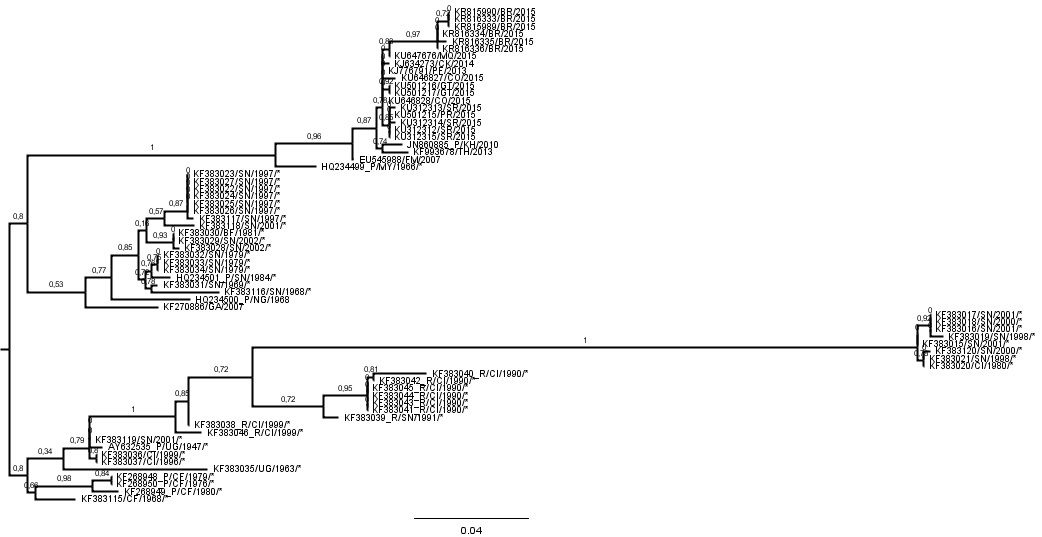
We did the ML tree for the ZIKVCOMPLETE dataset and as you suggested there is a clade comprised by 8 sequences with anomalously long branches.
So, we inferred the nucleotide substitution rate of the ZIKVNON-HUMAN E genes using two new datasets: 1) ZIKVNON-HUMAN-A that included all ZIKV sequences retrieved from mosquitos and monkeys minus those 8 “anomalous” secuences; and 2) ZIKVNON-HUMAN-B that included the same sequences than ZIKVNON-HUMAN-A minus those previously characterized as recombinant in the original Faye et al. (2014) publication.
Despite the new rate estimations are between 1.0-1.5 ×10-3 , as Dr. Rambaut observed before, the interval spans more than an order of magnitude. Therefore, I assume that these sequences have poor temporary structure. On the other hand, I wonder if anyone knows which are the ancestral lineages of the recombinant strains characterized by Faye et al. (2014). I could not find this in that paper.
Below I place the Table with the clock estimates and the ML ZIKVCOMPLETE E gene tree.
Thanks Daiana, that’s useful. As I said, I do worry about the integrity of the Faye et al. data. In addition, there are some Senegalese sequences from the 2000s that are very close to Uganda 1947. I’m sure this is not correct either.
I agree with Eddie - if you want a good, reliable, rate for the Americas outbreak, wait for more data from the outbreak. For the sylvatic virus in Africa then sampling over similar timescales would be good. It would be great to have genomes from the old viruses but relies on someone having a sample in the freezer somewhere.
One Senegalese sequence from 2001 is really very close to the prototype Uganda 1947, as well as two sequences from the Ivory Coast from 1996 and 1999. Maybe the problem is the prototype strain that accumulates many mutations as consequence of multiple passages. If that was the case, however, we should assume that was a convergent evolution with some of the recent African sequences isolated 50 years later. We can remove that Ugandan prototype sequence and estimate the molecular clock again. But if the problem is the integrity of the Faye et al data there is not much that we can do.
There is a way to calibrate the ZIKV molecular clock with human sequences while minimizing the potential problem of deleterious mutations at the terminal branches? Maybe only considering mutations at the internal branches of the tree?