Hi there,
I’m not a virologist but I was interested to have a quick look and see if there were any noticeable biases in the sequencing technology used for the nCoV-2019 genome sequences available at GISAID. Four different sequencing technologies have been used - Illumina, BGI, Nanopore and Sanger. Three different combinations of these four have also been employed by the various sequencing teams. This provides an interesting opportunity to see how the different platforms behave on this type of genome.
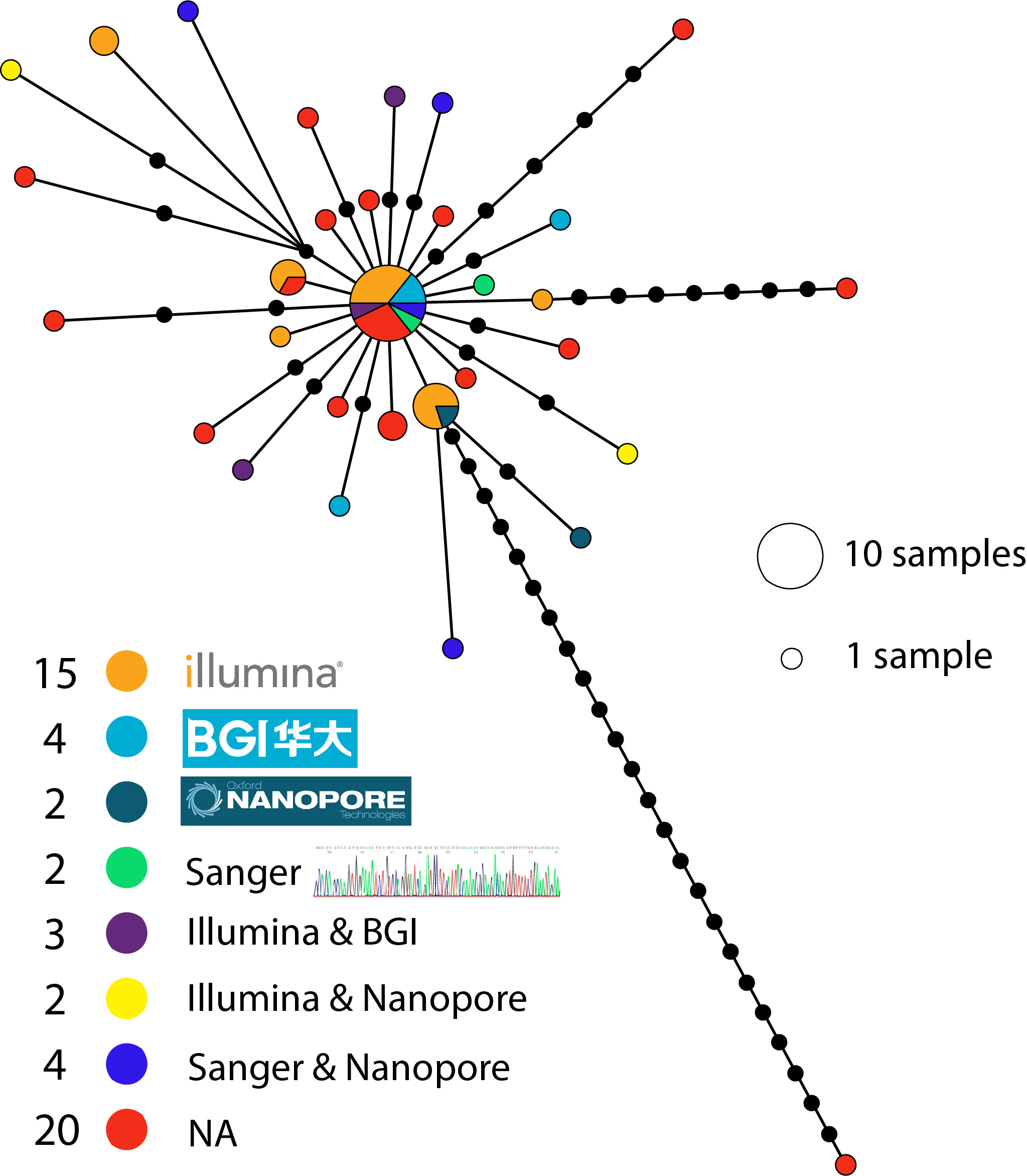
I downloaded the sequences and aligned them with MAFFT and clipped the ends of the alignment to the shortest sequence. I then made a tcs haplotype network in popart using the sequencing technology listed in the GISAID metadata to colour the haplotypes:
I also did the same analysis but using country instead of sequencing technology to try to see where in the world the different technology is being used (but perhaps some of the samples may have been sequenced in a different country to the one they were collected in).
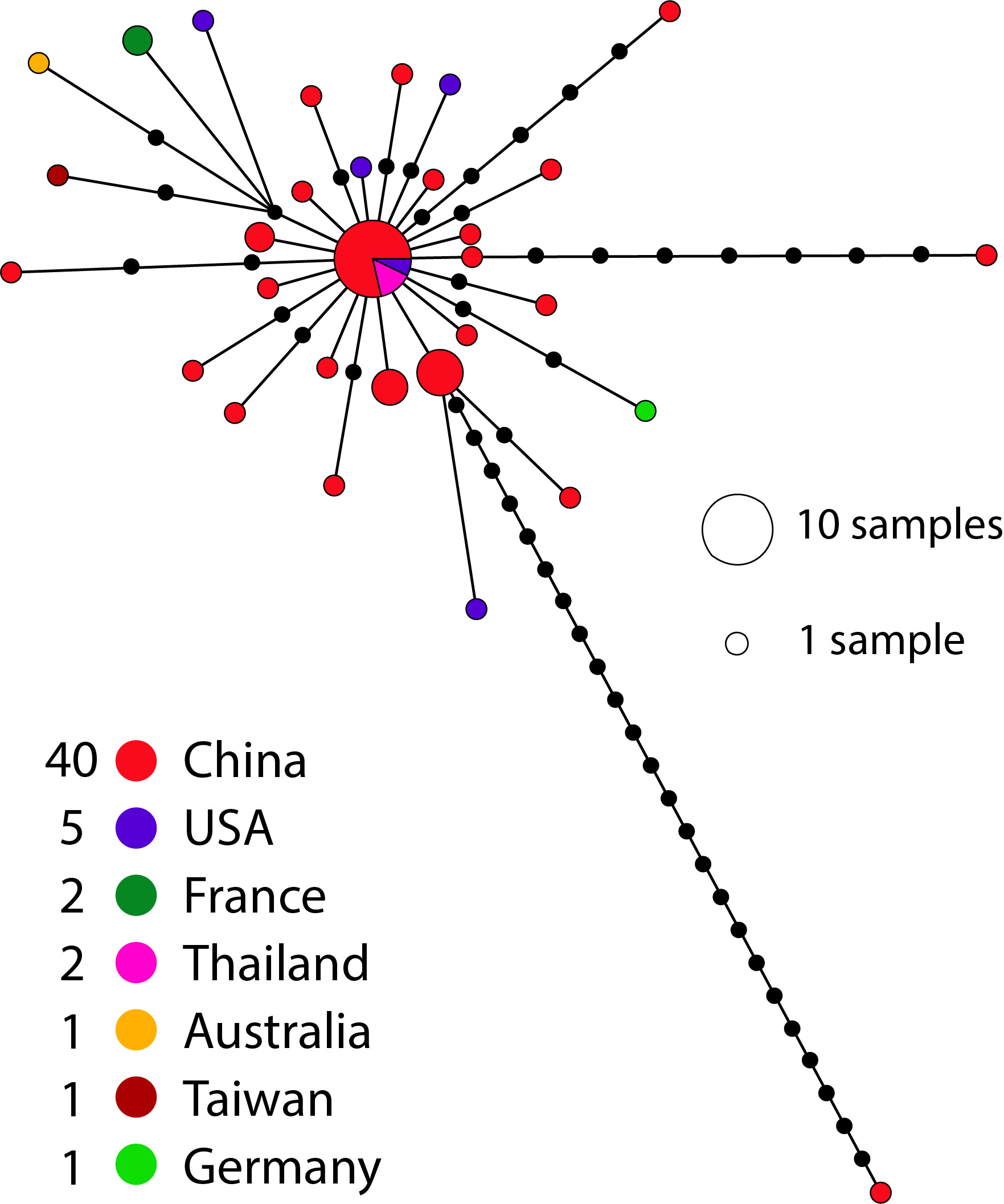
Most genomes have been sequenced on illumina machines (15 out of 52), with four sequenced by BGI machines. Overall there doesn’t seem to be any strong pattern of bias indicating that most of these technologies seem to be performing well on this genome. There are a few (three) outliers that seem to be due to errors, but the sequencing technology wasn’t provided in the metadata for them. The largest haplotype has been sequenced by all technologies with the exception of nanopore (although it was recovered in a Nanopore + Sanger assembly). Some of these patterns will become more clear as the number of genomes and different technologies increases.
I thought it was an interesting quick look at the various sequencing technologies and strategies and how they are performing.
I gratefully acknowledge the Authors, the Originating and Submitting Laboratories for their sequence and metadata shared through GISAID, on which this research is based. https://www.gisaid.org/