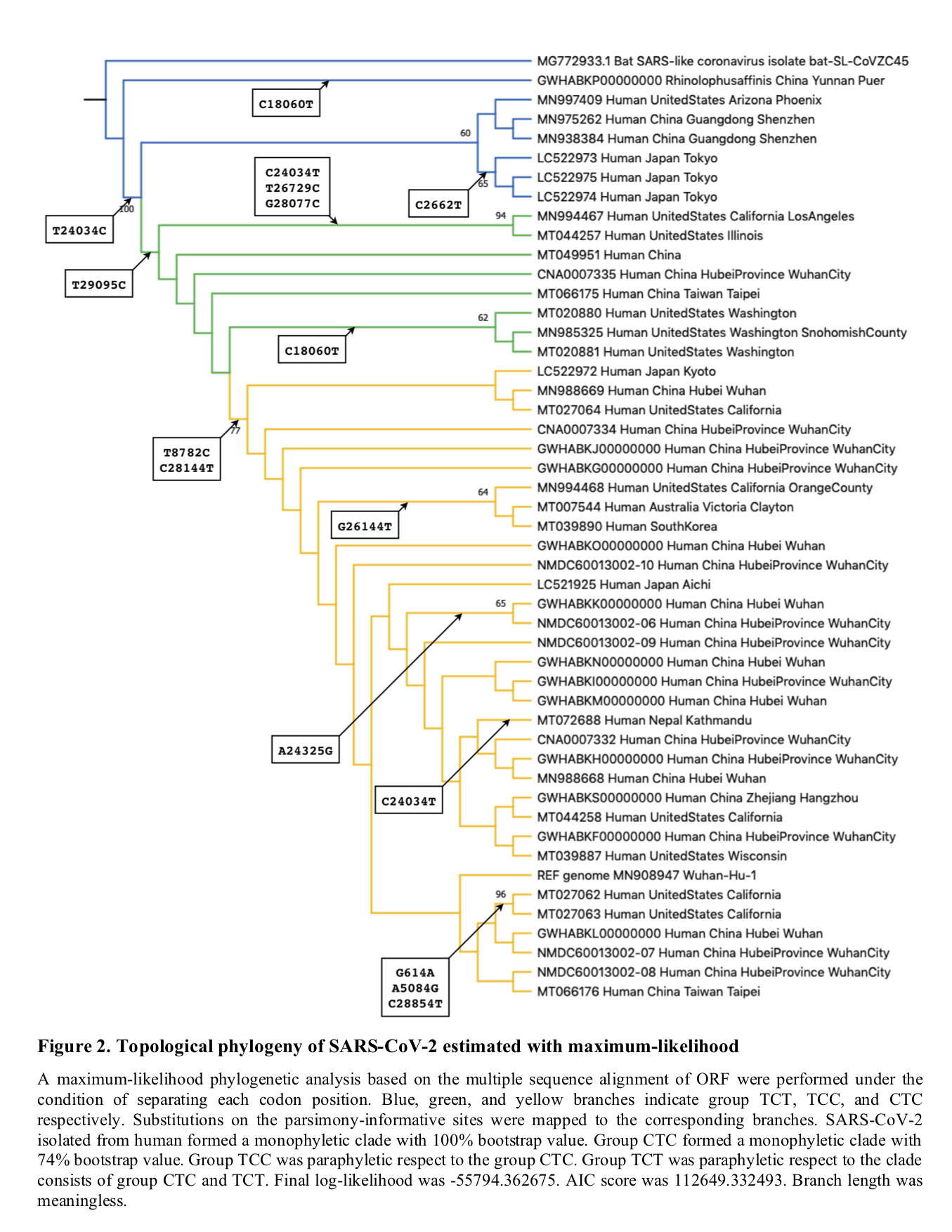
In our phylogenetic analyses, “L type” likely formed a monophyletic derived clade. “L type” in Tang et al. is same to “Group CTC” in our phylogenic tree.