Recent successes in sequencing Ebola and Zika viruses using MinION sequencing technology (Quick et al, 2016; Faria et al, 2017, Quick et al, 2017) have proved that high quality whole viral genomes can be generated in near real-time (often < 2 working days) during viral outbreaks. The portability, low cost-per-run, and real-time data production afforded by the MinION means that genomic data can be generated at the source of the outbreak in real time, permitting rapid channeling of the data to inform intervention strategies.
On 6th January 2017, Brazil reported a Yellow Fever Virus outbreak that started in December 2016 (ECDC 2017). The outbreak has since grown to be the largest observed in the country for decades, and has reached areas where vaccine coverage is limited. Until 20th April 2017, 1449 suspected and confirmed cases of yellow fever have been reported in Brazil, of which approximately half of all cases occurred in the state of Minas Gerais (ECDC, 2017). The outbreak has spread throughout much of the state, but most cases (including those confirmed by qPCR, viral isolation and also via serological positives) are concentrated in two distinct areas (Figure 1). Despite this extremely high case count, only two genomes have been published from the entire outbreak to date (Bonaldo et al, 2017) and none from Minas Gerais.
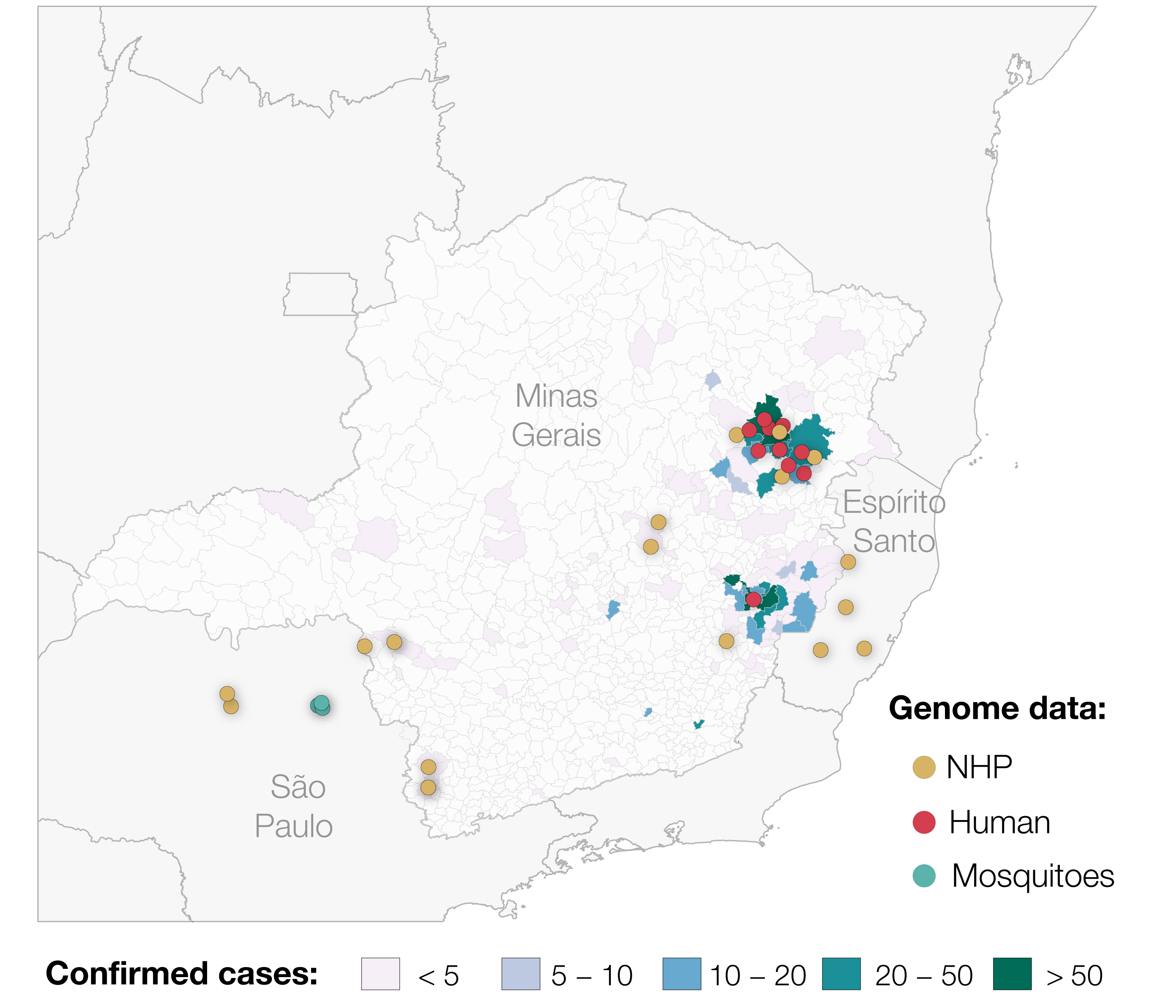
Figure 1. Map showing confirmed cases of Yellow Fever Virus in Minas Gerais and the sampling locations of genomes generated for this study (n=29, up to the 2 May 2017). We also include the locations of the 2 samples from Espirito Santo previously published by Bonaldo et al. 2017. In total, 793 cases confirmed by qPCR, virus isolation and/or serology at Fundação Ezequiel Dias in Belo Horizonte are shown.
Following recent success in sequencing of Zika virus on the minION using an overlapping PCR amplicon scheme generated by PrimalScheme (Quick et al, 2017), we employed a similar strategy for YFV. The 500bp primer scheme (which we have already made publically available here) was tested on the vaccine strain 17D at Public Health England, UK, prior to the work in Brazil. During a one week testing and sequencing marathon at FUNED, the Laboratório Central de Saúde Pública (LACEN) in Minas Gerais, teams of researchers from FUNED and other institutions in Brazil and the UK worked together to confirm positive qPCR results for ~90 YFV cases from primates and humans and perform PCR amplification assays on these samples. Using Nanopore MinION sequencing we were able to generate 20 full YFV genomes 7 days after arrival (up until 02 May 2017 - the data analysed here is thus provisional). 19 of the genomes are from fatal cases (including both humans and non-human primates). We are currently preparing further successfully amplified samples for sequencing within the next week.
Approximately two thirds of qPCR cases in our current set of samples for which cDNA synthesis and PCRs have been performed are from human cases (tissue and serum/plasma) and one third are from non-human primates (exclusively tissue samples from the genera Alouatta (family Atelidae), Callithrix (family Callitrichidae), and individuals from the family Cebidae, including Callicebus). The median qPCR cycle threshold values (Cts) for positive samples are notably lower in tissue than serum samples, with median Cts of 15.1 for tissue and 32 for blood products (ranges; 6.0-36.8 and 7.0-37.0).
We have constructed a maximum likelihood phylogenetic tree of our preliminary whole genome YFV data (n=20 full-length genomes), and we have included additional data from primates and mosquitos generated using Illumina MiSeq in Instituto Adolfo Lutz and the University of São Paulo (n=9). We have also included the only two publically available sequences from the outbreak, published by Bonaldo et al, 2017 (Figure 2). All samples generated by us cluster together in the tree along with the two previously generated genomes (Bonaldo et al, 2017) that were analysed together with several other partial genomes from the South American genotype 1 in an earlier virological post. Our data reveals two basal clades composed entirely of primates and Haemagogus mosquitoes, strongly suggesting a primate reservoir for the current outbreak. The earliest primate sequence is from early November 2016. A third clade of mixed human and primate cases suggests multiple subsequent spillovers into humans, as would be expected for Yellow Fever Virus circulating as part of a sylvatic cycle.
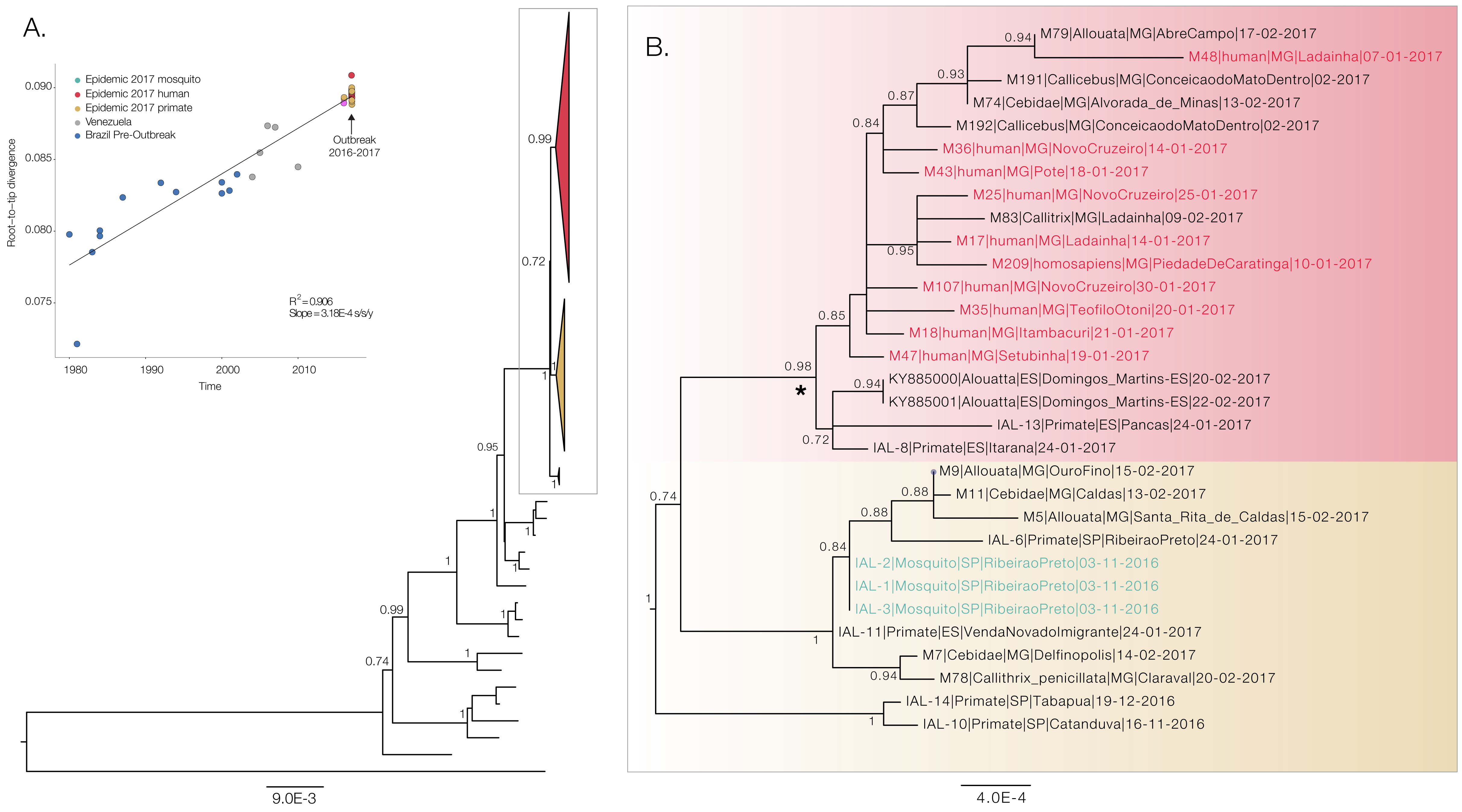
Figure 2. Maximum likelihood phylogeny of the South American clade 1 (SA1) genotype that is responsible for the current outbreak in Southeast Brazilian region (panel A). The inset shows the correlation between genetic distance from the tips to the root of the tree and sampling dates. Panel B shows a ML tree generated with PhyML under a GTR+G+I substitution model generated entirely with sequences from this outbreak. This includes sequences generated by us, and the two previously published sequences from Espírito Santo published by Bonaldo et al. 2017. Taxa names starting with M = genomes generated using Nanopore MinION sequencing by us during the last week at FUNED, Belo Horizonte, Brazil. IAL = genomes generated at Instituto Adolfo Lutz, São Paulo, Brazil. Asterisk indicates the outbreak clade that includes human fatal cases.
The strong temporal signal (Figure 2A insert) indicates that evolutionary clock models are appropriate for inferring evolutionary origins of this outbreak and suggests lack of mislabeling or contamination. We ran a preliminary evolutionary analysis using BEAST, under a GTR+G+I substitution model and a relaxed uncorrelated lognormal clock as in Bryant et al. 2007. The time of the most recent common ancestor of the YFV SA1 outbreak clade in Brazil containing human sequences (see Figure 2B) dates back to late Oct 2015 (95% Bayesian Credible Interval: Sep 2014 to Jul 2016, see Figure 3).

Figure 3. Posterior probability density for the estimated age of the most recent common ancestor of YFV SA1 clade with human cases in Minas Gerais, Brazil
Please note also that the results presented here should be considered preliminary. Additional analyses are currently being performed, and hypotheses about the ignition and spread of the outbreak will be interpreted in light of extensive epidemiological and ecological data. More to come soon!
References:
Quick et al., Real-time, portable genome sequencing for Ebola surveillance, Nature 2016 (available here)
Quick et al., Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples, 2017 Nature Protocols in press (BiorXiv version available here)
Faria et al., Epidemic establishment and cryptic transmission of Zika virus in Brazil and the Americas, Nature in press 2017 (BiorXiv version available here)
Bonaldo et al. Genome analysis of yellow fever virus of Brazil ongoing outbreak reveals polymorphisms, 2017 Mem Inst Oswaldo Cruz, in press (available here)
Outbreak of yellow fever in Brazil, 2017: Epidemiological situation, European Centre for Disease Control (available here)
Bryant et al., Out of Africa: A Molecular Perspective on the Introduction of Yellow Fever Virus into the Americas, PLoS Pathogens 2007 (available here)
Contributing authors by institution:
Sarah C Hill, Julien Thezé, Oliver G Pybus, Nuno Rodrigues Faria from the Department of Zoology, University of Oxford, United Kingdom
Jaqueline Goes de Jesus, Joilson Xavier, and Luiz Carlos Júnior Alcântara from the Centro de Pesquisa Gonçalo Moniz, Fundação Oswaldo Cruz, Salvador, Bahia, Brazil
Felipe Campos de Melo Iani, Talita Emile Ribeiro Adelino, Maira Alves Pereira, Glauco Carvalho, Marluce Aparecida Assunção Oliveira from the Central Laboratory of Public Health of Minas Gerais, Brazil, Octávio Magalhães Institute, Ezequiel Dias Foundation, Minas Gerais, Brazil.
Lívia Sacchetto, Izabela Maurício de Rezende, Poliana de Oliveira Figueiredo, Érica Munhoz de Mello, Betânia Paiva Drumond, Giliane de Souza Trindade, and Rodrigo Dias de Oliveira Carvalho from the Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
Moritz U. G. Kraemer from the Harvard Medical School & Boston Children’s Hospital, USA and from the Department of Zoology, University of Oxford, United Kingdom
Simon Cauchemez from the Institute Pasteur, Paris, France
Renato S Aguiar from the Universidade Federal do Rio de Janeir, Departamento de Genetica, Instituto de Biologia.
Nick Loman, Josh Quick from the Institute of Microbiology and Infection, School of Biosciences, University of Birmingham, Birmingham, United Kingdom
Renato Pereira de Souza, Fabiana Cristina Pereira dos Santos, Mariana Sequetin Cunha, Juliana Silva Nogueira, Iray Maria Rocco, Leandro Guariglia D’Agostino, Adriana Yurika Maeda, Fernanda Gisele da Silva Vasami e Fernando Luiz de Lima Macedo from the Núcleo de Doenças de Transmissão Vetorial, Instituto Adolfo Lutz, São Paulo, Brazil
Antonio Charlys da Costa, Ester Cerdeira Sabino from the Department of Infectious Disease, School of Medicine & Institute of Tropical Medicine, University of São Paulo, Brazil
Shirley Cavalcante Vasconcelos Komninakis from the Retrovirology Laboratory, Federal University of São Paulo, Brazil, School of Medicine of ABC (FMABC), Clinical Immunology Laboratory, Santo Andre, Brazil
Rosa Maria Tubaki, Regiane Maria Tironi de Menezes, Eduardo Sterlino Bergo from the Superintendência do Controle de Endemias - SUCEN, São Paulo, Brazil
Roberta Spinola from the Centro de Vigilância Epidemiológica “Prof. Alexandre Vranjac”, SES, São Paulo, Brazil
[submitted on behalf of Sarah Hill]