Updated on 2019.02.27
In 2018, two Zika outbreaks were reported from Northeast India: Rajasthan and Madhya Pradesh states. From both, 100-200 cases were reported, and the peak in October to November is similar to the peaks during the chikungunya epidemics in India ~10 years earlier (Figure 1).
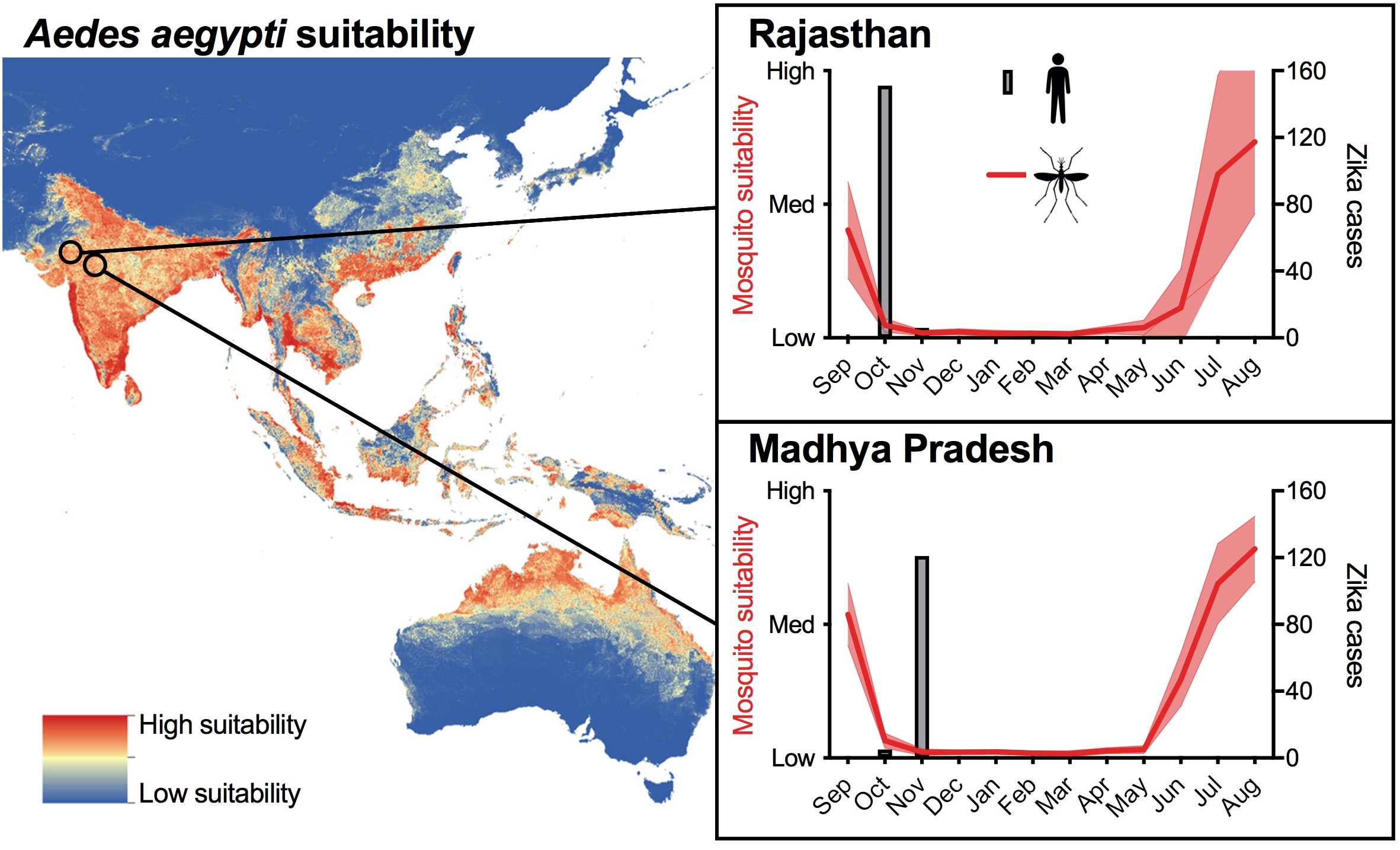
Figure 1. Overview of 2018 Zika outbreaks in India. _Aedes aegypt_i suitability map from Kraemer et al., and the temporal suitability data was curated by Moritz Kraemer and Alex Watts. Zika virus case numbers are from the Madhya Pradesh government and news reports. We made a version of this for The Hindu.
Yadav et al. recently published 5 complete or near-complete genomes from the 2018 Zika virus outbreak in Jaipur (Rajasthan state). Their findings show that the viruses belong to a lineage that is endemic to Asia and that did not spread through the Pacific and Americas. However, there is still much that we do not know, including: (1) when the outbreak in Rajasthan started, (2) where the virus came from, and (3) if the outbreak in Madhya Pradesh is related.
Currently, we are collaborating with Profs. Sarman Singh and Debasis Biswas from the All India Institute of Medical Sciences to sequence and analyze Zika virus from the Madhya Pradesh outbreak, and are seeking collaborators to sequence addition clinical samples from the region (including from travelers). Hopefully, we will have results from that data soon.
To start, we did preliminary analysis with the five Yadav et al. Zika virus genomes and another available genomes from the Americas and Asia extracted from Virus Variation database. We aligned a total of 57 genomes using MAFFT.
For the phylogenetic analyses, we used jModelTest to determine the best fit model of substitution, which was revealed to be GTR+I+G. Using PhyML 3.0, we inferred an initial Maximum Likelihood (ML) tree by applying Subtree Pruning and Regrafting (SPR) algorithm for topology searching, with branch supports calculated using a SH-like approximate likelihood-ratio test (aLRT).
To investigate how clock-like the molecular data are, we performed root-to-tip analysis of the ML tree using Tempest (Figure 2C). To infer the time-calibrated (Figure 2A), we used a Markov Chain Monte Carlo (MCMC) Bayesian approach implemented in BEAST v1.10.4, using a relaxed molecular clock model, along with a Bayesian SkyGrid model as tree prior. An analysis using SkyGrid and strict clock priors was also performed, but the selected model (SkyGrid + relaxed clock) was the best fitting one, showing the highest marginal likelihood estimated using path-sampling and stepping-stone approaches. Our analysis reached convergence after 30 million generations, and its results were summarized as a maximum clade credibility (MCC) tree using TreeAnnotator.
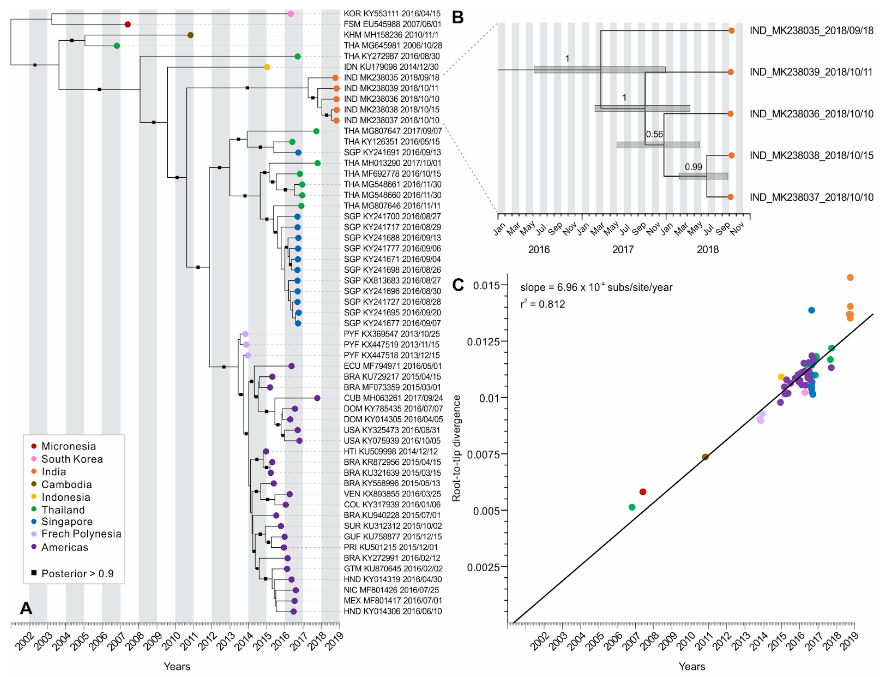
Figure 2. Evolution of Zika virus from an outbreak in Rajasthan, India, 2018. (A) Maximum clade-credibility phylogenetic tree of Zika virus genomes from the Americas and Asia (including five recently released Indian genomes). Branch supports are given as posterior probabilities, shown as black squares along the branches with posterior > 0.9. (B) Inset zooming in on the Zika virus genomes sequenced from Rajasthan, India. Bars at internal nodes depict the 95% highest posterior density (HPD) interval for divergence times, which indicate March 2017 as the tMRCA of this Zika virus lineage. (C) Root-to-tip regression analysis performed using a ML tree of the Zika virus genomes shown in (A). A strong linear relationship was observed between sequence divergence (subs/site) and time (years) (r2 = 0.812), with genomes evolving under similar rates of ~6.96 × 10−4 nucleotide substitutions per site per year (subs/site/year).
A PDF of Figure 2 can be downloaded here: 20190226-Zika_India-virologicalFig2.pdf (420.6 KB)
Our analyses using the Yadav et al. Zika virus genomes show that indeed the outbreak in Rajasthan was caused by a virus endemic to Asia, but we cannot currently determine exactly where it came from or if it is endemic to India (Figure 2A) - it connects to viruses sampled from throughout Southeast Asia by a very long branch. We are pursuing addition sequencing in the region to help fill in some of these gaps.
We estimated that the tMRCA of the Jaipur (Rajasthan state) clade is March, 2017, however, with a wide confidence interval (from June, 2016 to December, 2017; Figure 1B). Still, it appears that local Zika virus transmission leading to the outbreak in Rajasthan started 1-2 years prior to the observed peak in cases (October, 2018), indicating that ‘silent’ transmission was occuring in the region. Zika virus had been reported in India during this period, including Gujarat in November 2016 (3 cases) and Tamil Nadu in June 2017 (1 case). Again, whether these are small, unconnected outbreaks caused by endemic Zika viruses or the result of a single recent emergence into region requires further investigation.
We are also looking for additional collaborators, especially who would like help sequencing Zika viruses from India or the surrounding areas, so please let us know if you are interested.
Please let us know if you have any questions or concerns about our analyses, and we will provide updates as we acquire more data. The alignment, skyline plots, and tree files are available at: GitHub - grubaughlab/project_2019_ZIKV-india: Preliminary analysis of the Zika virus outbreaks in India.
All credit for the Zika virus genomes sequenced from India presented here should go to:
Pragya, D. Yadava, Bharati Malhotrab, Gajanan Sapkala, Dimpal A. Nyayanita, Gururaj Deshpandea, Nivedita Guptac, Ullas T. Padinjaremattathila, Himanshu Sharmab, Rima R. Sahaya, Pratibha Sharmab, Devendra T. Mouryaa
Zika virus outbreak in Rajasthan, India in 2018 was caused by a virus endemic to Asia
Anderson Brito and Nathan Grubaugh
Department of Epidemiology of Microbial Diseases
Yale School of Public Health