Phylodynamic analysis of a densely sampled COVID19 outbreak in Weifang, China
Erik M Volz, Han Fu 1 , Haowei Wang 1 , Xiaoyue Xi 2 , Wei Chen 3 , Dehui Liu 3 , Yingying Chen 3 , Mengmeng Tian 3 , Wei Tan 4 , Junjie Zai 5 , Wanying Sun 6 , Jiandong Li 6 , Junhua Li 6 , Xingguang Li 7, Qing Nie 3
- Department of Infectious Disease Epidemiology and MRC Centre for Global Infectious Disease Analysis, Imperial College London,2.Department of Mathematics, Imperial College London,3. Department of Microbiology, Weifang Center for Disease Control and Prevention, Weifang, 4. Department of Respiratory Medicine, Weifang People’s Hospital, Weifang, 5. Immunology Innovation Team, School of Medicine, Ningbo University, Ningbo,6. Shenzhen Key Laboratory of Unknown Pathogen Identification, BGI-Shenzhen, Shenzhen, 7. Hubei Engineering Research Center of Viral Vector, Wuhan University of Bioengineering, Wuhan, China
We report an analysis of 20 whole SARS-CoV 2 genomes from a single relatively small and geographically constrained outbreak in the city of Weifang in Shandong Province, People’s Republic of China. The outbreak in Weifang took place far from the epicentre of the Chinese epidemic in Wuhan and was rapidly brought under control in early February. These data are unique in coming from a single location and an outbreak that was quickly controlled. Thus the 20 sequences represent a substantial proportion of all indidviduals ever infected in Weifang.
These data were released on the same day as a large batch of sequences from Guangdong described by Oliver Pybus here. Together, these datasets have increased the number of sequences from China by about 70%. The sequence data are released on GISAID: EPI_ISL_413691, EPI_ISL_413692, EPI_ISL_413693, EPI_ISL_413694, EPI_ISL_413695, EPI_ISL_413696, EPI_ISL_413697, EPI_ISL_413711, EPI_ISL_413729, EPI_ISL_413746, EPI_ISL_413747, EPI_ISL_413748, EPI_ISL_413749, EPI_ISL_413750, EPI_ISL_413751, EPI_ISL_413752, EPI_ISL_413753, EPI_ISL_413761, EPI_ISL_413791, EPI_ISL_413809
We prepared a preprint describing these data and a phylodynamic analysis which can be found here. Below I provide a summary of the main results and discuss some methodological issues. Feedback would be welcome regarding application of model-based phylodynamic analyses of datasets with low genetic diversity. Given the short period of this outbreak and low overall diversity, the data present speical challenges for interpreting Bayesian analyses as the choice of prior can have huge influence on results.
Epidemiological investigation, sampling, and genetic sequencing
The following details are from our preprint: As of 10 February 2020, 136 suspected cases, and 214 close contacts were diagnosed by Weifang Center for Disease Control and Prevention. 28 cases were detected positive with SARS-CoV-2. Viral RNA was extracted using Maxwell 16 Viral Total Nucleic Acid Purification Kit (Promega AS1150) by magnetic bead method and RNeasy Mini Kit (QIAGEN 74104) by column method. RT-qPCR was carried out using 2019 novel coronavirus nucleic acid detection kit (BioGerm, Shanghai, China) to confirm the presence of SARS-CoV-2 viral RNA with cycle threshold (Ct) values range from 17 to 37, targeting the high conservative region (ORF1ab/N gene) in SARS-CoV-2 genome. Metagenomic sequencing: The concentration of RNA samples was measurement by Qubit RNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). DNase was used to remove host DNA. The remaining RNA was used to construct the single-stranded circular DNA library with MGIEasy RNA Library preparation reagent set (MGI, Shenzhen, China). Purified RNA was then fragmented. Using these short fragments as templates, random hexamers were used to synthesize the first-strand cDNA, followed by the second strand synthesis. Using the short double-strand DNA, a DNA library was constructed through end repair, adaptor ligation, and PCR amplification. PCR products were transformed into a single strand circular DNA library through DNA-denaturation and circularization. DNA nanoballs (DNBs) were generated with the single-stranded circular DNA library by rolling circle replication (RCR). The DNBs were loaded into the flow cell and pair-end 100bp sequencing on the DNBSEQ-T7 platform 8 (MGI, Shenzhen, China). 20 genomes were assembled with length from 26,840 to 29,882 nucleotides. The median age of patients was 36 (range:6-75). Two of twenty patients suffered severe or critical illness. Weifang sequences were combined with a diverse selection of sequences from China outside of Weifang and other countries provided by GISAID.
Phylogenetic analysis
We aligned the 20 Weifang sequences using MAFFT with a previous aligment of 35 SARS-CoV2 sequences from outside of Weifang used here. Maximum likelihood analysis was carried using IQTree with a HKY+G4 substitution model and a time-scaled tree was estimated using treedater 0.5.0. Two outliers according to the molecular clock model were identified and removed using `treedater’ which was also used to compute the root to tip regression.
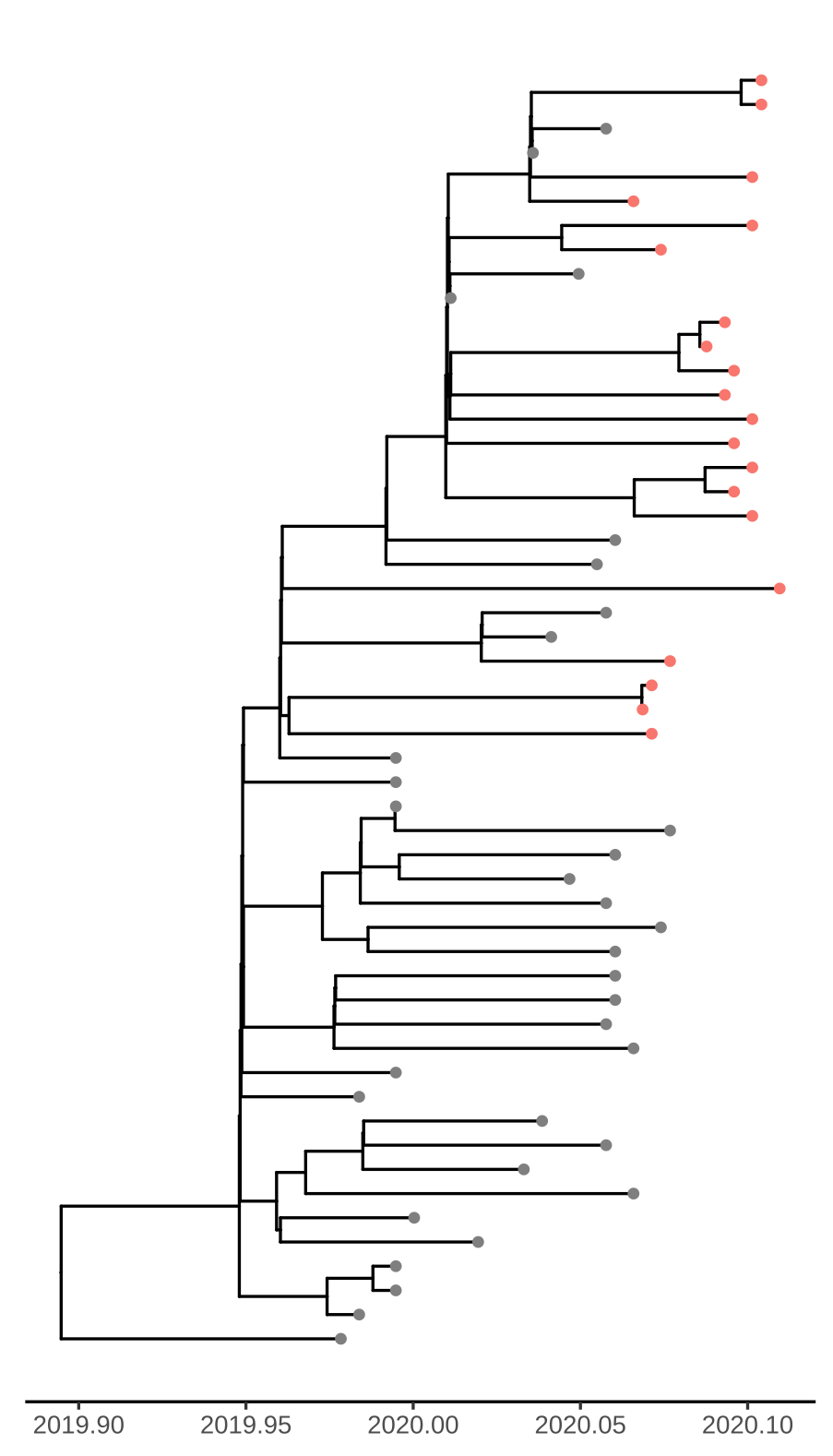
Figure 1. Time scaled tree estimated by maximum likelihood and treedater. Samples from Weifang are shown in red.
Addition of the Weifang sequences greatly improves clock signal in comparison to what was reported in the MRC Centre Report 5 and appears similar to the RTT presented here.
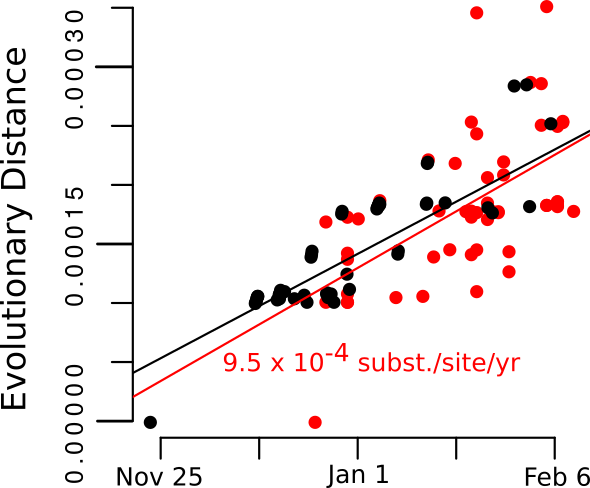
Figure 2. Root to tip regression plot.
Model-based phylodynamic analysis
We used a compartmental SEIR model implemented in BEAST2 PhyDyn package to estimate R0, cases through time, and TMRCAs. Choice of this model was guided by the following design goals:
- We must account for importations (potentially more than one) into Weifang from a much larger international reservoir
- We must account for nonlinear dynamics in Weifang. We want the model to have the potential to detect a nonlinear decrease in cases (although we will see that it did not)
- In order to estimate cases through time, we want to
- Realistically model generation times by including a non-infectious incubation period
- Account for high variance in transmission rates which is known to influence thee relationship between Ne and population size (cf discussions here)
The equations describing dynamics in Weifang are as follows:
$$
\dot{S}(t) = - \beta \left( \beta I(t) + \beta \tau J(t) \right) \frac{S(t)}{S(t) + I(t) + J(t) + R(t)} \
\dot{E}(t) = \beta \left( \beta I(t) + \beta \tau J(t) \right) \frac{S(t)}{S(t) + I(t) + J(t) + R(t)} - \gamma_0 E(t) \
\dot{I}(t) = \gamma_0 (1-p_h) E(t) - \gamma_1 I(t) \
\dot{J}(t) = \gamma_0 p_h E(t) - \gamma_1 J(t) \
\dot{R}(t) = \gamma_1 (E(t) + J(t) )
$$
S: Susceptbles, E: incubation period, I:infectious, and R: recovered. In these equations, J represents a minority compartment (20%) with much higher transmission rate. The proportion and infectiousness of J is chosen to give an overdispersion of the offspring distribution similar to what was observed in the 2003 SARS epidemic.
Outside of Weifang, we say that the epidemic is growing exponentially, and we define a constant per-lineage bidirectional rate of migration between Weifang and the international reservoir.
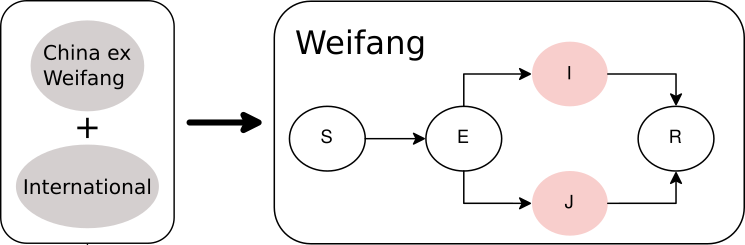
Figure 3. Diagram of SEIJR model fitted in BEAST. Colours correspond to tips of the trees.
The time scaled phylogeny estimated in BEAST shows dramatically different lineages through time than the dated ML tree showing the strong influence of the model-based prior.
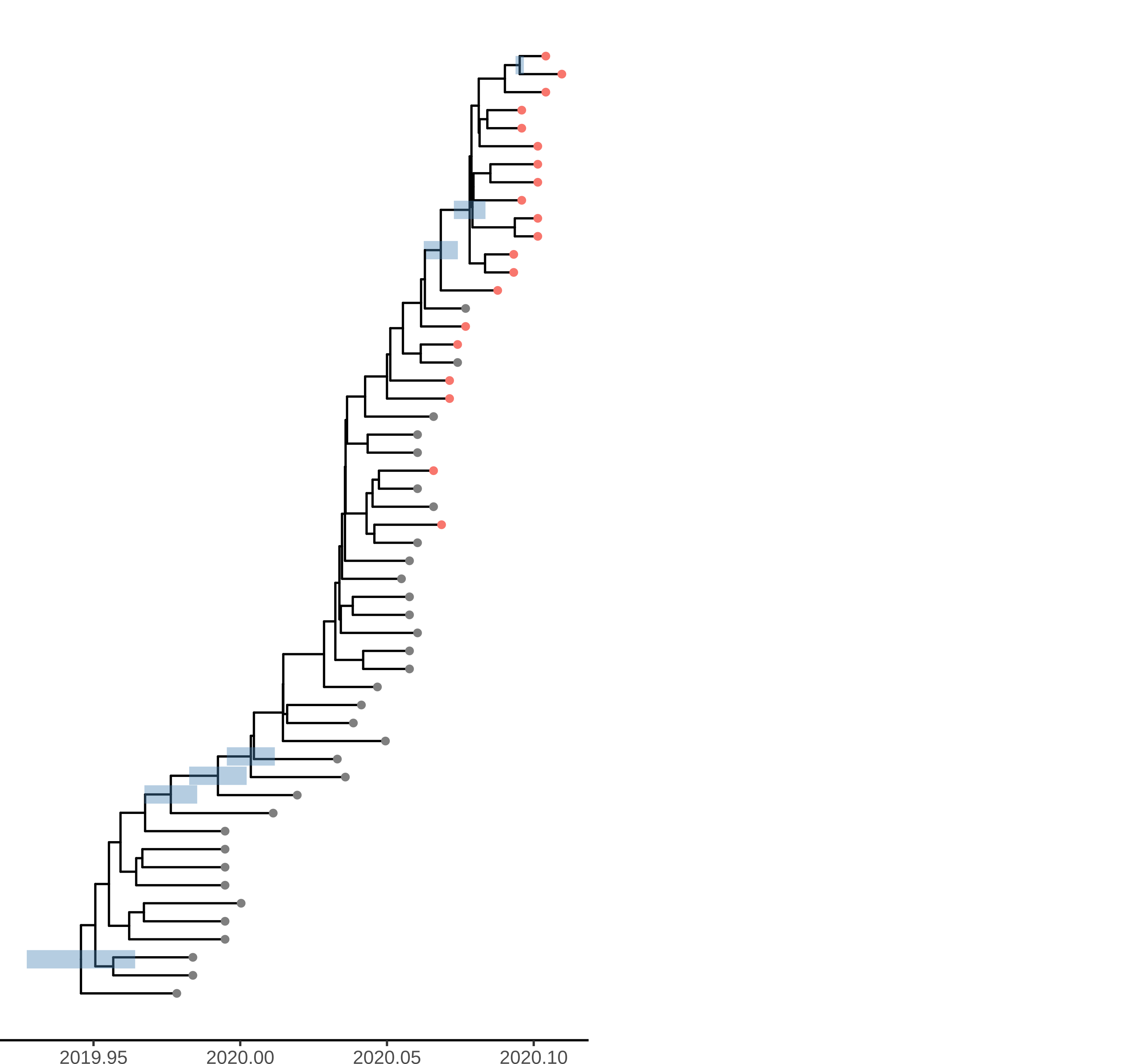
Figure 4. Time scaled phylogeny estimated in BEAST with the SEIJR model
Unsurprisingly very few nodes have strong posterior support. The phylogeny shows HPD of TMRCA for nodes with >50% support. Among these are 3 nodes ancestral to Weifang sequence. The preprint also shows a densitree plot. We can tentatively conclude that there were relatively few introductions in to Weifang. The TMRCA of this clade likely occurred Jan 15-Jan 23. These dates overlap with the first sample from Weifang: The earliest Weifang sequence was sampled on January 25 from a patient who showed first symptoms on January 16.
The Weifang outbreak was quickly brought under control and maximum confirmed cases occurred on February 5. Unfortunately, our sequence data only extend to February 10 and we are not able to detect a subsequent decline.
Here we show the estimated number of cases estimated with the SEIJR model in comparison to confirmed cases. Note that we should expect estimated infections to exceed confirmed cases because
- An unknown proportion of cases will have asymptomatic or mild illness which makes them difficult to detect
- There is a lag between infection and appearance of symptoms and a lag from symptoms to diagnosis. Exceptions can occur if mild/asymptomatic cases are detected by contact tracing.
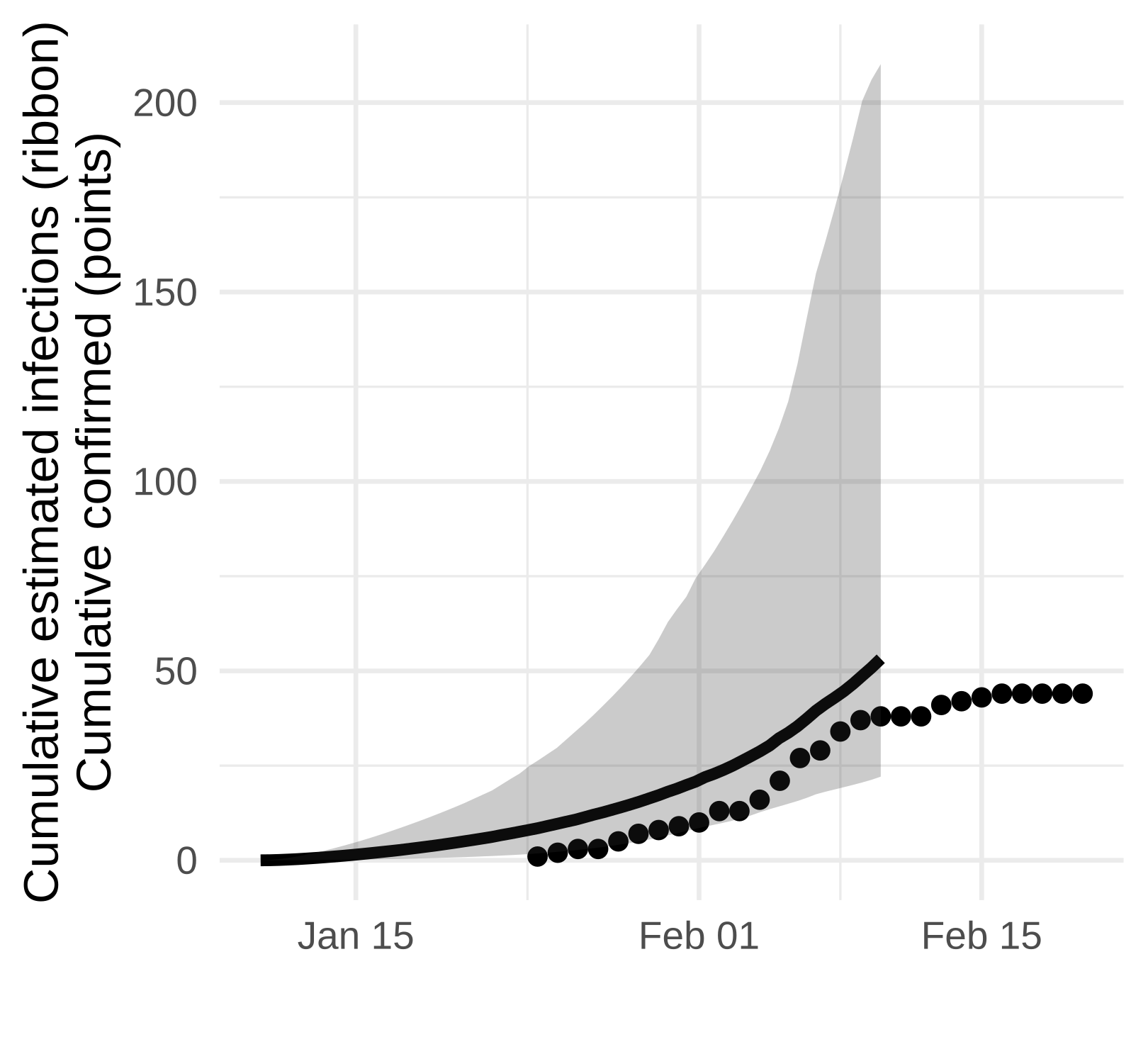
Figure 5. Estimated confirmed cases (points) and estimated cumulative infections (line and ribbon)
While we don’t detect a dramatic slow down in growth, the estimated number of cases is consistent with the number reported up to the end of February and this analysis suggests that there is not a very large hidden burden of infection within this particular city.
Finally, we of course estimate R0 from the SEIJR model: 1.99(95% CI:1.48-3.14). This is a bit lower than estimated in other settings, but this makes sense in light of the fact that the outbreak took place over a period when a variety of public health interventions were being deployed.
Open questions
The priors for the epidemiological parameters in this model were mostly uninformative except for those that describe natural history of infection and variance in transmission rates which were fixed. Nevertheless:
- How sensitive are R0 and case estimates to the parametric form of the epidemiological model?
- Would incorporation of more international sequences influence results?
Suggestions for future modeling studies would be welcome.
Code to replicate this analysis and and BEAST XML files can be found at
https://github.com/emvolz/weifang-sarscov2.
Acknowledgements
These analyses would not be possible without the enormous efforts and transparency of outbreak investigators at Weifang CDC and scientists at BGI-Shenzhen and Wuhan University of Bioengineering.
These groups are open to future collaborations and wish to maximize the scientific impact of their data:
We ask that you communicate with Xinguang Li([email protected]) & Qing Nie if you wish to publish results that use these data in a journal. If you have any other questions, please also contact us directly.
This work was supported by Centre funding from the UK Medical Research Council under a concordat with the UK Department for International Development. NIHR. J-IDEA. This work was also supported by a grant from the Special Project for Prevention and Control of Pneumonia of New Coronavirus Infection in Weifang Science and Technology Development Plan in 2020 (2020YQFK015) to Associate Senior Technologist Qing Nie.
Role of the Funders: All funders of the study had no role in study design, data analysis, data interpretation, or writing of the report.
We gratefully acknowledge China National GeneBank at Shenzhen, China for the sequencing strategy and capacity support. We also gratefully acknowledge the laboratories that have contributed publicly available genomes via GISAID: Shanghai Public Health Clinical Center & School of Public Health, Fudan University, Shanghai, China, at the National Institute for Viral Disease Control and Prevention, China CDC, Beijing, China, at the Institute of Pathogen Biology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China, at the Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan, China, at the Department of Microbiology, Zhejiang Provincial Center for Disease Control and Prevention, Hangzhou, China, at the Guangdong Provincial Center for Diseases Control and Prevention at the Department of Medical Sciences, at the Shenzhen Key Laboratory of Pathogen and Immunity, Shenzhen, China, at the Hangzhou Center for Disease and Control Microbiology Lab, Zhejiang, China, at the National Institute of Health, Nonthaburi, Thailand, at the National Institute of Infectious Diseases, Tokyo, Japan, at the Korea Centers for Disease Control & Prevention, Cheongju, Korea, at the National Public Health Laboratory, Singapore, at the US Centers for Disease Control and Prevention, Atlanta, USA, at the Institut Pasteur, Paris, France, at the Respiratory Virus Unit, Microbiology Services Colindale, Public Health England, and at the Department of Virology, University of Helsinki and Helsinki University Hospital, Helsinki, Finland, and at the University of Melbourne, Peter Doherty Institute for Infection and Immunity, Melbourne, Australia, at the Victorian Infectious Disease Reference Laboratory, Melbourne, Australia, at the Public Health Virology Laboratory, Brisbane, Australia and at the Institute of Clinical Pathology and Medical Research, University of Sydney, Westmead, Australia.