Re-emergence of Zika virus in Africa: circulation of the Asian lineage Zika virus in Angola
Angola and Cape Verde have recently reported both the presence of Zika virus and a near-simultaneous increase in the number of cases of microcephaly detected nationally. Genetic data from these events are limited to a single, short sequence from a neonate with microcephaly born in Portugal to a resident of Angola (Sassetti et al. 2018: First case of confirmed congenital Zika syndrome in continental Africa - PubMed). Due to the very short subgenomic region sequenced (193 bp), analysis of this fragment from the NS5 gene can only deliver an incomplete picture of the ZIKV diversity circulating in Angola (Thézé et al., 2018: Genomic Epidemiology Reconstructs the Introduction and Spread of Zika Virus in Central America and Mexico - PubMed). Moreover, the lack of complete genome sequences from Africa makes it impossible to fully understand the re-emergence and extent of ZIKV spread within the continent.
We have previously established successful protocols for amplifying ZIKV genomes using an overlapping PCR amplicon scheme and for sequencing ZIKV amplicons on the Nanopore MinION (Quick et al, 2017: Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples - PMC). For two weeks in the summer of 2018, a team of researchers from the UK, Brazil and Angola congregated at the Instituto Nacional de Investigação em Saúde (INIS), Ministry of Health, in Luanda, the capital city of Angola, and worked together to sequence ZIKV positive samples from Angola. The team was able to successfully sequence two samples in Luanda that had been previously identified as positive for ZIKV during routine surveillance activities conducted at the INIS. Subsequently, we successfully recovered a third ZIKV genome at the Instituto Nacional de Saúde Doutor Ricardo Jorge (Portuguese Ministry of Health), from a newborn with microcephaly whose mother was infected in Angola. This case has been previously reported alongside 193bp of sequence data (Sassetti et al. 2018), but through additional sequencing we were able to successfully recover the majority (82%) of the genome. A summary of the epidemiological information regarding each sequenced strain, and the proportion of the coding region that was successfully sequenced at a depth of >20 bases, is described below:
- Patient Z3 : Sample was collected on the 23 May 2017 from a female patient residing in the Bengo province. The patient reported no travel in the weeks before onset of symptoms. Sequence coverage was 75%.
- Patient P153 : Sample collected in June 2017 from a HIV+ female patient residing in Luanda reported no travel and no symptoms. Sequence coverage was 85%.
- Patient PoHuZV/634959 : sample was recovered from a baby born in November 2017 with microcephaly to a mother that was infected in the first trimester/early second trimester. We refer to Sassetti et al. 2018 for clinical and epidemiological details. Sequence coverage was 82%.
Maximum likelihood and Bayesian phylogenetic analysis of ~400 ZIKV sequences indicate that all three sequenced genomes from Angola belong to the Asian genotype and form a single well-supported monophyletic clade (bootstrap = 100, posterior probability support = 0.93, Figure 1a and b ). Importantly, our analyses indicate that the Angolan ZIKV clade shares a common ancestor with lineages circulating in Brazil, possibly in the southeast region of the country where nearly 3000 microcephaly cases have been reported since 2015 (posterior probability = 0.88). Our genetic data therefore suggest that ZIKV may have been introduced to Angola from Brazil, with possible endemic circulation in Angola prior to the first reports of Zika virus circulation in Dec 2016 (http://www.who.int/emergencies/zika-virus/situation-report/20-january-2017/en/). However, it is also possible that ZIKV was introduced to Angola via an as-yet unsampled location, or location from which identified viruses have not yet been sequenced, such as Capo Verde). The median estimated date for the most recent common ancestor of the Angolan sequences was August 2016 (95% HPD May - November 2016). The median estimated date for the most recent common ancestor linking this clade with non-Angolan sequences was August 2015 (95% HPD December 2014 to December 2015). This suggests that ZIKV may have been first introduced to Angola no earlier than ~August 2015, and no later than ~August 2016.
We are currently conducting further analyses to better explore the direction and timing of the introduction of Asian-lineage ZIKV in Angola.
Between March 2017 and June 2018, 50 samples were taken from babies born with microcephaly in Angola ( Figure 1c ). None of the samples taken from babies born in Angola with microcephaly were positive for the presence of ZIKV RNA by RT-qPCR, possibly because only 15/50 samples were taken within 1 week of birth, when RNA is most likely to be present. Note that the detected ZIKV RT-qPCR positive cases occur ~ 6 – 12 months prior to the peak of births of babies with microcephaly observed in Angola; given the low amount of available data, this is consistent with the time lag between ZIKV infection and microcephaly that has been observed in other countries, including Brazil (Faria et al. 2016: Zika virus in the Americas: Early epidemiological and genetic findings - PubMed) and Cape Verde (Lourenço et al. 2018: Epidemiology of the Zika Virus Outbreak in the Cabo Verde Islands, West Africa – PLOS Currents Outbreaks). We are currently analysing additional evidence of historical exposure to ZIKV from IgM and IgG ZIKV serological essays. We note that the strain of ZIKV identified in the microcephalic baby born in Portugal is closely phylogenetically clustered with other ZIKV in Angola, confirming that the strain identified here to be circulating endemically in Angola is associated directly with microcephaly. Our findings strongly suggest an association between the reported spike in microcephaly in Luanda and the circulation of Zika virus in the country.
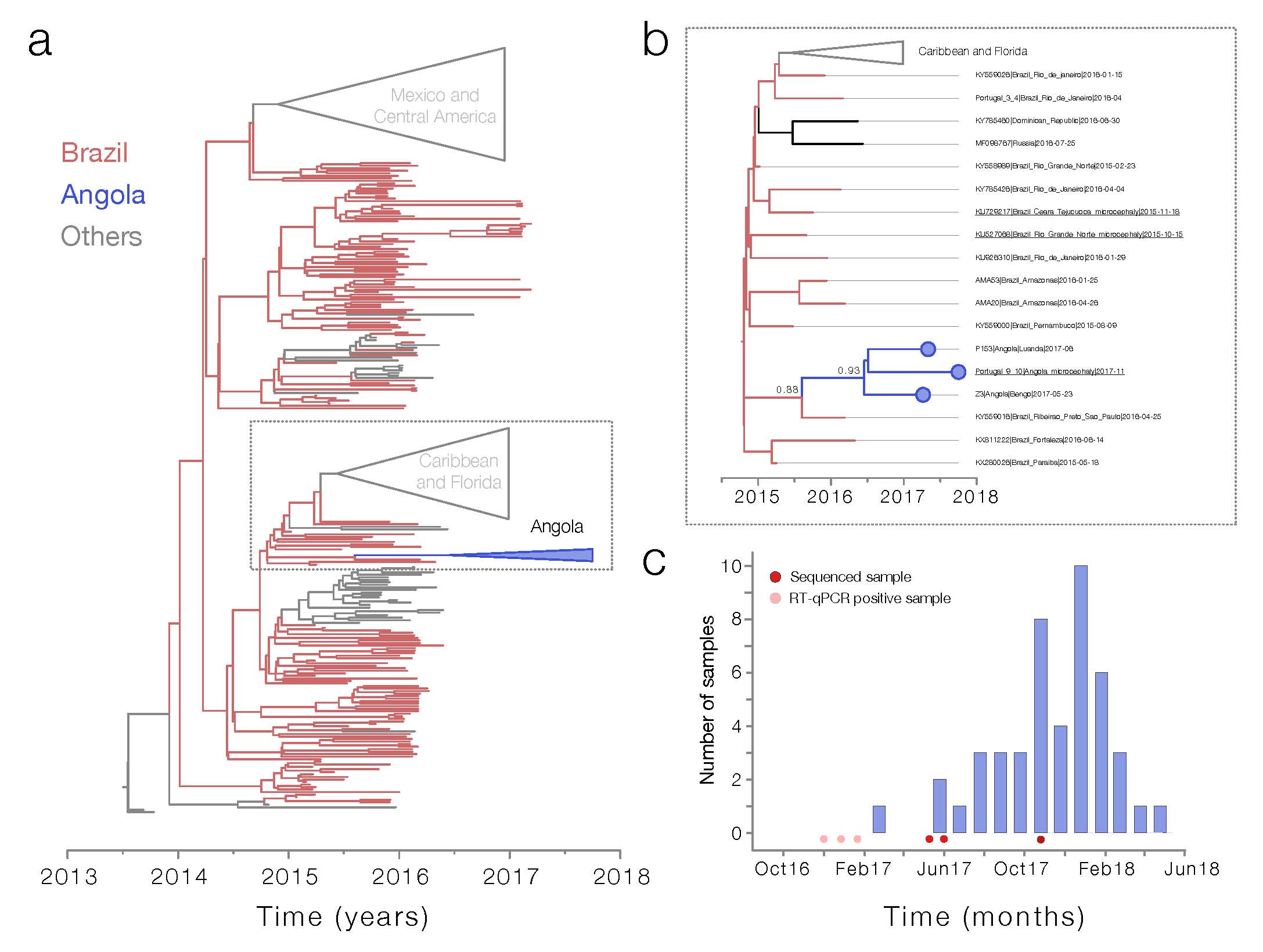
Figure : Zika virus and associated microcephaly curve in Angola. Panel a shows the maximum likelihood phylogeny of the complete genomes (n=380) from Zika virus Asian genotype. The clade containing the Angolan sequences is shown in bold, and expanded into panel b . Support for branching structure is shown by bootstrap values at nodes. Node bars at key nodes show the 95% highest posterior density intervals of the node age. Angolan sequences are highlighted in red. Panel c shows the number of babies with microcephaly per month reported to INIS.
Acknowledgments:
This work was funded by a Wellcome Trust and Royal Society Sir Henry Dale Fellowship (204311/Z/16/Z), by an internal GCRF-HEFCE (005073) and by a John Fell Research Fund (grant 005166) from the University of Oxford, UK. Travel to Angola was supported by grants from AfOx (AfiOX-60). Julien Thézé and Oliver Pybus were supported by the ERC grant Pathphylodyn. This work was supported by the Oxford Martin School.
Authors :
Sarah C. Hill, Department of Zoology, University of Oxford, U.K.
Jocelyne Vasconcelos, Instituto Nacional de Investigação em Saúde, Ministry of Health, Luanda, Angola.
Líbia Zé-Zé, Instituto Nacional de Saúde Doutor Ricardo Jorge, Águas de Moura, Portugal and Biosystems and Integrative Sciences Institute (BioISI), Edificio TecLabs, Portugal.
Zoraima Neto, Molecular Biology Laboratory, Instituto Nacional de Investigação em Saúde, Ministry of Health, Luanda, Angola.
Domingos Jandondo, Molecular Biology Laboratory, Instituto Nacional de Investigação em Saúde, Ministry of Health, Luanda, Angola.
Marinela Mirandela, Molecular Biology Laboratory, Instituto Nacional de Investigação em Saúde, Ministry of Health, Luanda, Angola.
Cruz dos Santos Sebastião, Molecular Biology Laboratory, Instituto Nacional de Investigação em Saúde, Ministry of Health, Luanda, Angola.
Ana Luísa Micolo Cândido, Molecular Biology Laboratory, Instituto Nacional de Investigação em Saúde, Ministry of Health, Luanda, Angola.
Carina Clemente, Cligest Clinic, Luanda, Angola.
Sara Pereira da Silva, Cligest Clinic, Luanda, Angola
Renato Santana Aguiar, Universidade Federal do Rio de Janeiro, Brazil.
Joilson Xavier, FioCRUZ Rio de Janeiro, Brazil.
Julien Thézé, Department of Zoology, University of Oxford, U.K.
Luiz Carlos Junior Alcantara, FioCRUZ Rio de Janeiro, Brazil.
Ingra Morales, Instituto de Medicina Tropical e Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil.
Kamran Khan, Department of Medicine, University of Toronto, Canada and Li Ka Shing Knowledge Institute, St Michael’s Hospital, Toronto, Canada
Alexander G. Watts, Li Ka Shing Knowledge Institute, St. Michael’s Hospital, Toronto, Canada
Domingos Quibuco, Hospital Pediátrico David Bernardino, Luanda, Angola
Cristóvão Domingos, Instituto Nacional de Luta Contra SIDA, Luanda, Angola
Bárbara Pocongo, Instituto Nacional de Luta Contra SIDA, Luanda, Angola
Ester C. Sabino, Instituto de Medicina Tropical e Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil.
Oliver G. Pybus, Department of Zoology, University of Oxford, U.K.
Maria-João Alves, Instituto Nacional de Saúde Doutor Ricardo Jorge, Águas de Moura, Portugal.
Joana Afonso, Instituto Nacional de Investigação em Saúde, Ministry of Health, Luanda, Angola.
Nuno R. Faria, Department of Zoology, University of Oxford, U.K.
Disclaimer
Please note though that these analyses are still based on work in progress and should be considered preliminary. Our analyses of this data are ongoing and a publication communicating our findings on these and other published genomes is in preparation. If you intend to use these data or would like access to the newly generated sequences from Angola prior to our publication, please contact us directly to coordinate.
Nuno Rodrigues Faria (University of Oxford, UK): [email protected]
Joana Afonso (Ministry of Health, Angola): [email protected]