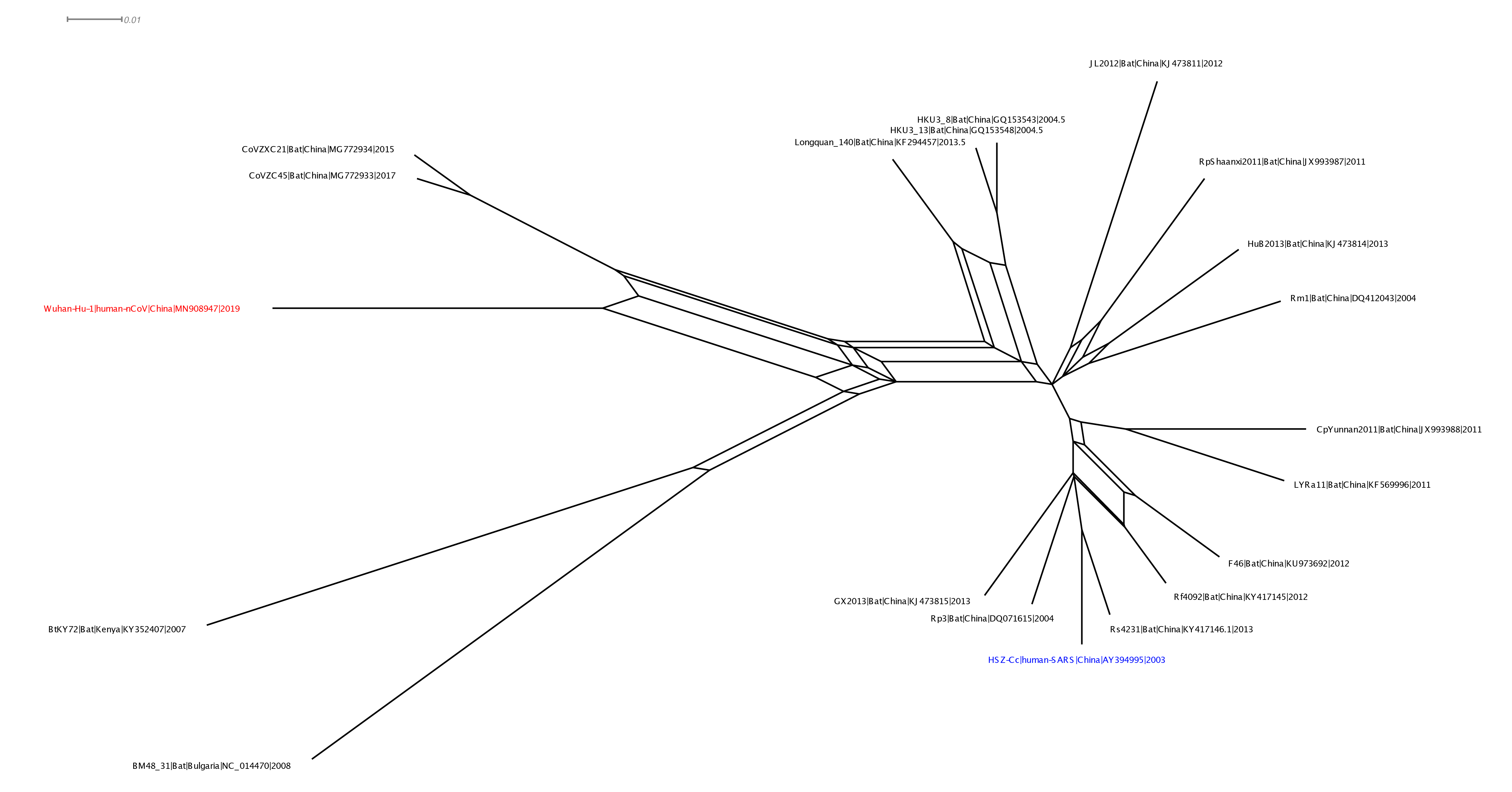
With Xiaowei Jiang at XJTLU we’ve carried out a preliminary evolutionary analysis to characterise the evolutionary origins of the Wuhan virus, nCoV. Focus of our analysis is on the Wuhan-Hu-1 virus (accession no. MN908947, released on GenBank by Shanghai Public Health Clinical Center and School of Public Health, Fudan University, Shanghai, China) as all nCoV cluster together so will share the same evolutionary ancestry. It’s clear from phylogenetic analysis the new human virus is most closely related to bat coronaviruses in the Betacoronaviruses genera. While this is apparent from both the previously reported BLAST and full-genome phylogenetic analysis the closest related bat viruses (CoVZC45 and CoVZXC21) are in fact recombinants with shared breakpoints either side of ORF1b:
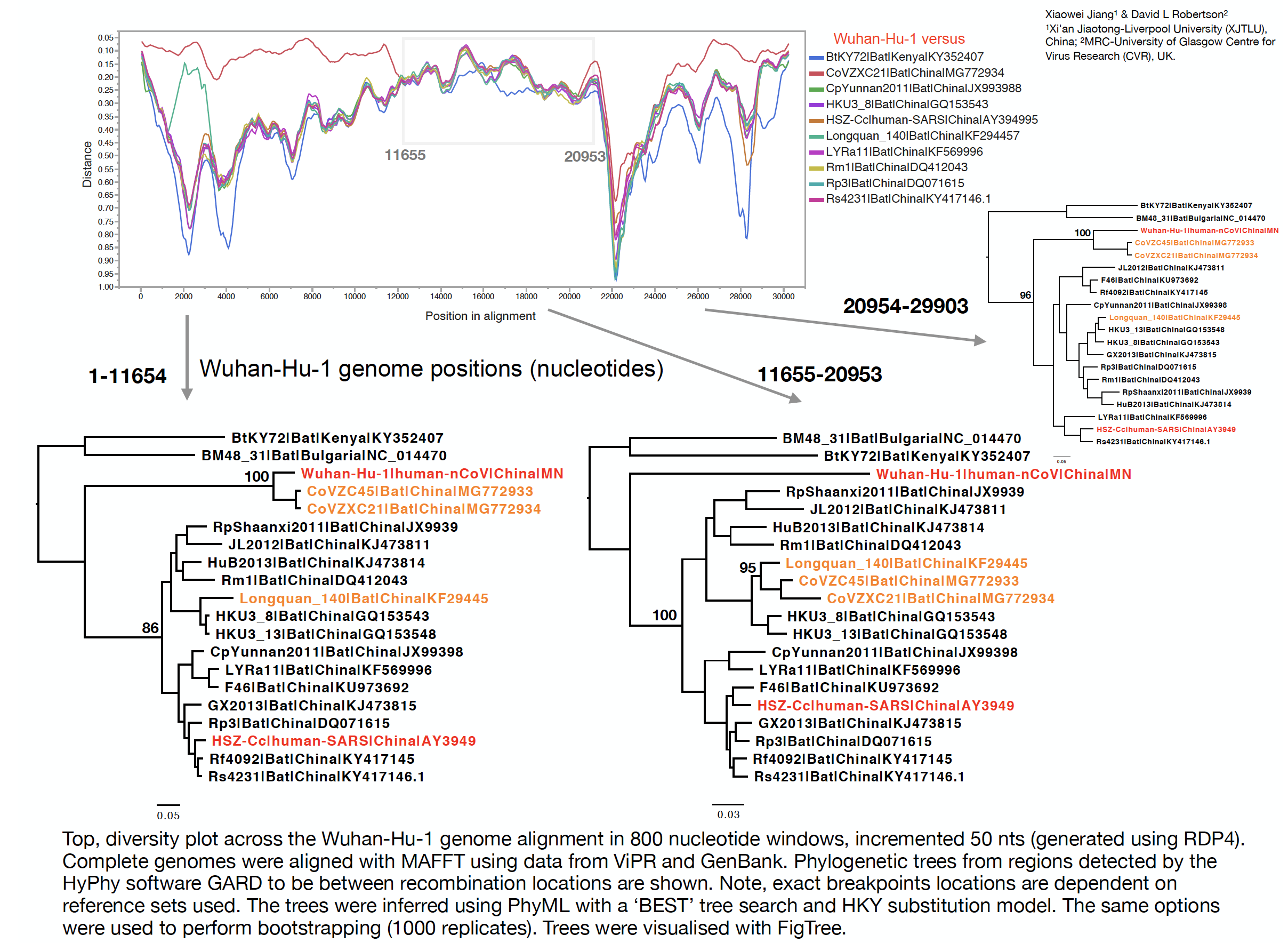
The phylogenetic clustering of the Wuhan-Hu-1 virus is consistently as a sister group to the SARS-related bat coronaviruses. Interestingly, a third bat coronavirus (Longquan_140) is a recombinant involving the Wuhan virus lineage in part of ORF1a.
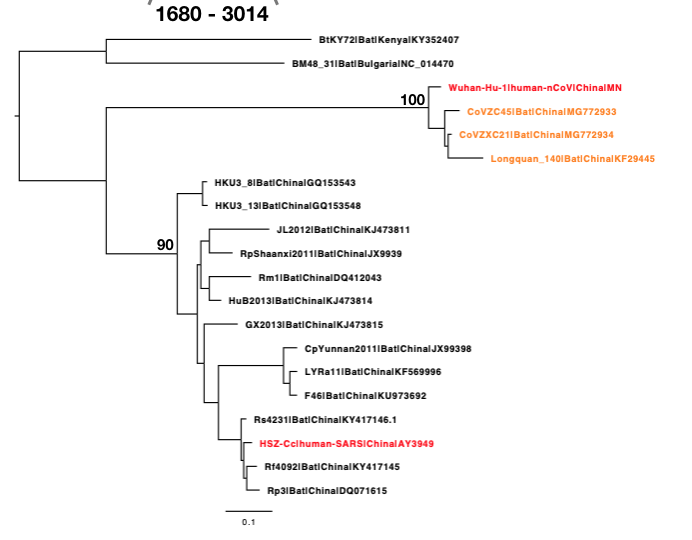
This analysis has detected three bat coronavirus recombinants (two with shared breakpoints) involving the nCoV lineage indicating greater diversity in the Chinese Sarbecovirus group than previously appreciated. The clustering of the related Sarbecovirus viruses from Kenya and Europe suggest the Wuhan virus is still part of the Sabecovirus sub-genre, and these recombination events probably occurred in bats. Although. given the propensity of coronaviruses to switch hosts, involvement of another species cannot be discounted. There is also a very good chance that a non-bat intermediate species is responsible for the beginning of the current outbreak in Wuhan. Given the tight clustering of the nCoV viruses in phylogenetic trees it seems most likely one event has occurred.
Several of these bat coronaviruses have been previously detected to be recombinant under-scoring the importance of doing appropriate analysis when analysing these viruses using phylogenetic methods. Recombination, in this case between divergent coronaviruses circulating in bats, violates our assumption of a single evolutionary tree and so needs to be considered carefully when inferring coronavirus evolution from complete genome alignments. We’re looking into the patterns of breakpoints to see if there’s any clues to the significance (or not) of these recombination events.
We’d like to thank the researchers and health professionals for making the nCoV data available. Credit also needs to be given to the surveillance projects for generating the data that is now available for comparison and to the software developers for making the tools we’ve used freely available: FigTree, available at: FigTree; GARD, available at http://www.hyphy.org; MAFFT, available at MAFFT - a multiple sequence alignment program; PhyML, available at ATGC: PhyML; and RDP4, available at ATGC: PhyML.