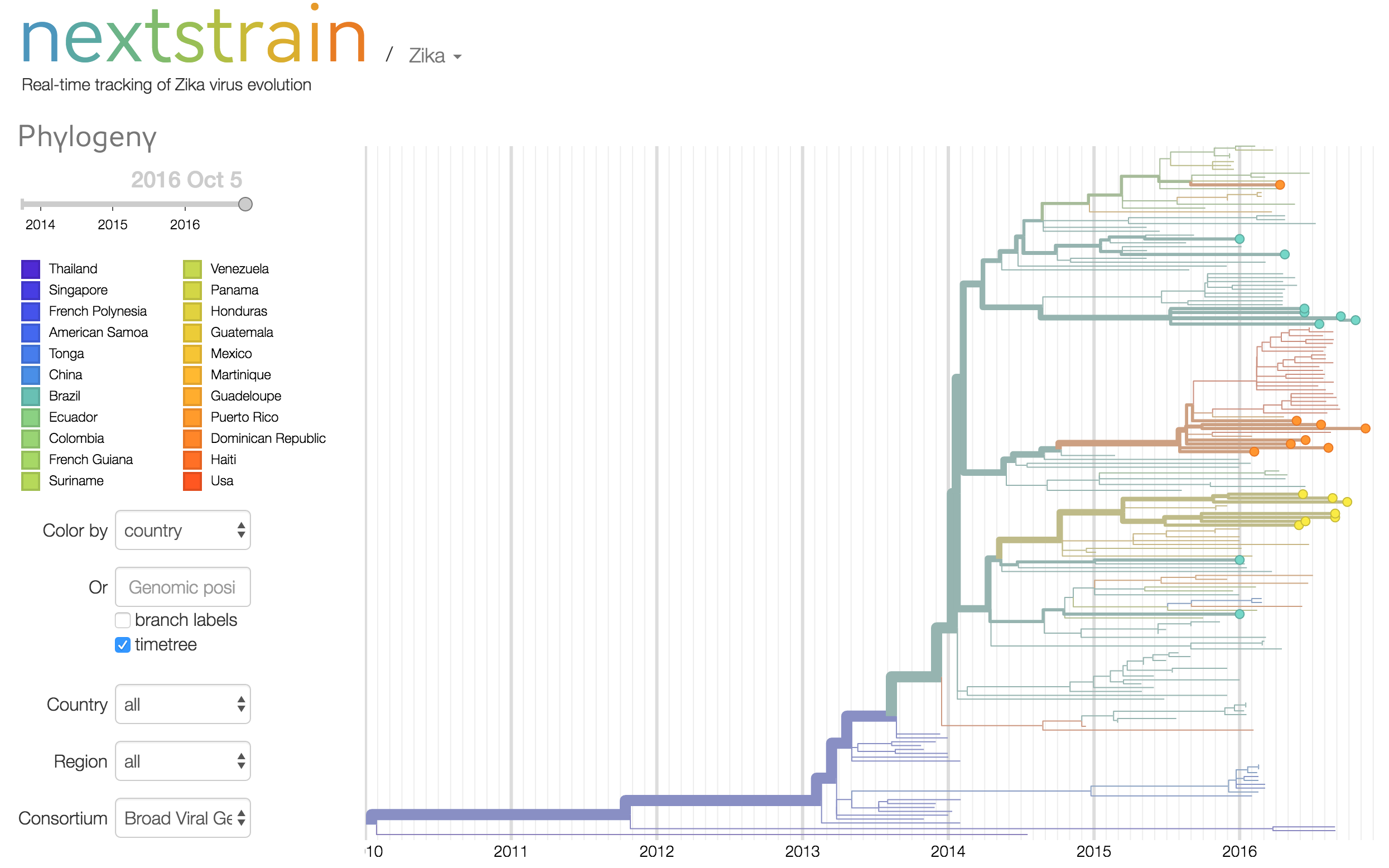
33 Zika virus genomes sequenced from patient and pooled mosquito samples collected in Rio de Janeiro, Tegucigalpa, Santo Domingo, and Florida, March-September 2016
The Broad Viral Genomics Group and MIT, together with partners at Oswaldo Cruz Foundation (Fiocruz) in Brazil; National Autonomous University of Honduras, and Florida Gulf Coast University (FGCU), are releasing 33 near-complete Zika virus (ZIKV) genomes sequenced from patient samples and mosquito pools collected in Brazil, Honduras, the Dominican Republic, and Florida between March and September 2016.
We are releasing the assemblies as consensus genomes and are in the process of depositing assemblies and read data, as well as sample metadata (generously shared by partners and including: place and date of collection, age, sex, and date of symptom onset), on NCBI GenBank.
The 33 ZIKV consensus sequences can be downloaded here:
ZIKV_BROAD_2016-10.fasta.txt (342.5 KB; this is a fasta file ending in ‘.txt’ because virological only allows certain file extensions)
Of the 33 samples, 9 are from Rio de Janeiro, Brazil, 7 are from Tegucigalpa, Honduras, 8 are from Santo Domingo, Dominican Republic, and 9 are from Florida, United States. 29 are from Zika-infected patients (10 urine, 7 plasma, 12 serum) and 4 are from ZIKV+ mosquito pools caught in Florida. All 33 genomes have >70% of the Zika genome and 22 have >95% of the Zika genome covered.
The samples were not cultured or passaged prior to sequencing. We experimented with different unbiased library preparation methods, and/or used amplicon-based sequencing, followed by Illumina sequencing on MiSeq and HiSeq platforms at the Broad Institute. Genomes were assembled using the Sabeti lab’s viral-ngs pipeline, which creates a de novo assembly from cleaned reads and performs additional refinement in a reference-assisted scaffolding process.
We assembled 17 Zika genomes after enriching libraries for Zika content by the Sabeti lab’s hybrid capture protocol. 13 have median coverage >25x and 4 have median coverage >250x. The other 16 were sequenced with amplicon-based sequencing using a protocol developed by the ZiBRA project and modified by the Andersen lab. We are still optimizing our pre-sequencing sample protocols, and will document our full process in a future update.
For more information about this project and data release, contact [email protected].
Disclaimer:
Please feel free to download, use, and share this data. Our partners and we are currently in the process of preparing publications describing our wet lab and genome analysis methods, and describing viral evolution and transmission patterns. We will post progress regarding data improvements, publications, and terms of use on this forum. If you intend to use these sequences for publication prior to the release of our papers, please contact us directly. If you are interested in joining our collaboration—or if you have any other questions—please also contact us directly.
The work was and continues to be part of a productive and growing collaboration across nations. Those currently involved include the following investigators and their teams:
Pardis Sabeti, Broad Institute and Harvard University, USA
Irene Bosch and Lee Gehrke, MIT, USA
Thiago Moreno L. Souza, Patrícia T. Bozza, Dr. Gonzalo Belo, and Wim Degrave, Oswaldo Cruz Foundation, Brazil
Fernando Bozza, Oswaldo Cruz Foundation and D’or Institute, Brazil
Fabiano L. Thompson, Federal University of Rio de Janeiro, Brazil
Kristian Andersen, The Scripps Research Institute, USA
Salim Mattar, Universidad de Córdoba, Colombia
Ivette Lorenzana, Universidad Nacional de Honduras, Honduras
Sharon Isern and Scott F. Michael, Florida Gulf Coast University, USA
Daniel Olson and Edwin J. Asturias, Colorado School of Public Health, USA

ZIKV_BROAD_2016-10_ML-TREE.pdf (332.7 KB)