Liu et al.1 reported that the infectivity of Zika virus (ZIKV) in Aedes aegypti mosquitoes was enhanced by an alanine-to-valine amino acid substitution at residue 188 of the NS1 protein. The authors suggest that the Asian lineage of ZIKV acquired enhanced infectivity when it spread from the Southeastern Asia to the Southern Pacific around 2013, because residue 188 of the NS1 protein was alanine in ZIKV isolates from the Asian clade collected before 2012, but was mutated to valine in all isolates collected after 2013. This hypothesis, however, was not formally tested using a model-based statistical framework.
Here, we performed a Bayesian evolutionary and phylogeographic analysis to reconstruct the spatiotemporal dissemination dynamics of the ZIKV Asian genotype and to properly estimate the age and location of the ancestral viral strain carrying the A188V substitution. Analysis of the complete coding regions of 45 ZIKV genome sequences from Southeastern Asia (n = 13, 1966-2016), Southern Pacific (n = 16, 2013-2016) and the Americas (n = 16, 2014-2015) reveals a very strong correlation (R2 = 0.99) between genetic divergence and sampling time within the ZIKV Asian lineage (Fig. 1a). The mean evolutionary rate (9.2 × 10-4 substitutions/site/year) and the overall time-scale of the Bayesian phylogenetic tree (Fig. 1b) here estimated for complete genomes of ZIKV Asian lineage were fully consistent with those previously reported2. Phylogeographic analyses using asymmetric (Fig. 1b) and symmetric (not showed) diffusion models placed the most recent common ancestor of ZIKV Asian genotype human strains in Southeastern Asia (posterior state probability, PSP = 1) at around 1999 [95% Bayesian credible interval (BCI): 1995-2003]. From Southeastern Asia, the ZIKV Asian genotype was disseminated to Pacific islands on two independent occasions. The first dissemination originated a large epidemic of ZIKV in Micronesia (Western Pacific) in 20073, but was not further spread to other countries. The second dissemination was dated to 2012 (BCI: 2012-2013) and fuelled recent ZIKV epidemics (2013-2016) in several Southern Pacific islands4,5. Our phylogeographic analyses supports a single entry of ZIKV from Southern Pacific (PSP = 1) into the Americas at 2013 (BCI: 2012-2013). Ancestral sequence reconstruction at internal nodes of the inferred ZIKV Asian genotype phylogeny place the emergence of the NS1 A188V substitution in Southeastern Asia (PSP = 1) at some time between 2003 (BCI: 2000-2006) and 2007 (BCI: 2004-2009) (Fig. 1b). In addition to the NS1 A188V and E V473M amino acid replacements described by Liu et al.1, our analysis detected four additional amino acid mutations (prM V1A, E T487M, NS3 N400H and NS4B M98I) that were concurrently fixed during dissemination of ZIKV in Southeastern Asia between 2003 and 2007, and two mutations fixed later when the virus spread to Southern Pacific (prM S17N) and the Americas (NS5 M114V).
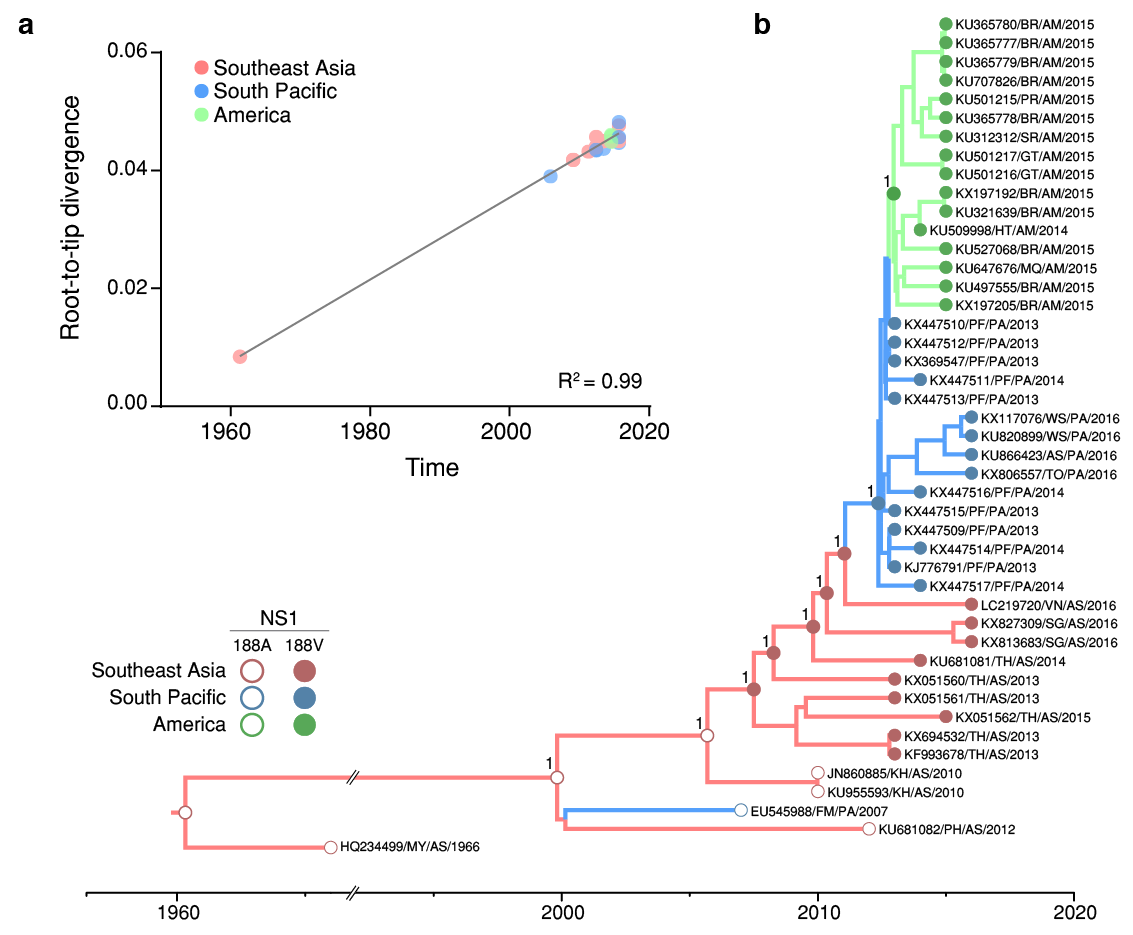
Figure 1 | Time-scale of the ZIKV NS1 A188V substitution emergence. a, There is a strong correlation (R2 = 0.99) between the sampling date of each sequence and the genetic distance of that sequence from the root of a maximum likelihood phylogenetic tree. Colours indicate the geographic region of sampling. b, Bayesian time-scale Maximum Clade Credibility phylogenetic tree estimated from ZIKV Asian genotype genomic sequences. Reconstructed ancestral key nodes and terminal nodes are highlighted according with the amino acid inferred or present at NS1 188 residue, respectively.
In summary, we showed that the NS1 A188V substitution associated with enhanced infectivity of ZIKV Asian lineage in Aedes aegypti mosquitoes probably arose during viral dissemination among human populations in the Southeastern Asian region, between early and middle 2000s. Thus, ZIKV Asian genotype strains carrying the NS1 A188V mutation appear to have spread in the Southeastern Asian region for some time (5-10 years) before being disseminated to Southern Pacific islands and the Americas. The absence of the reversal NS1 V188A mutation at either internal nodes or terminal tips in the ZIKV Asian genotype phylogeny clearly supports some selective advantage for the fixation of the valine amino acid at residue 188 in NS1. We further described six additional amino acid substitutions in other viral proteins that were also fixed during spread of ZIKV Asian lineage in the Southeastern region between early and middle 2000s, or after viral dissemination to the Southern Pacific and American regions. The potential impact of these additional mutations for ZIKV transmissibility and/or pathogenesis should be certainly explored.
References:
1 Liu, Y. et al. Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature, 2017. (available here)
2 Faria, N. R. et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science 352, 345-34, 2016. (available here)
3 Duffy, M. R. et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. The New England journal of medicine 360, 2536-2543, 2009. (available here)
4 Cao-Lormeau, V. M. et al. Zika virus, French polynesia, South pacific, 2013. Emerging infectious diseases 20, 1085-1086, 2014. (available here)
5 Musso, D., Nilles, E. J. & Cao-Lormeau, V. M. Rapid spread of emerging Zika virus in the Pacific area. Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases 20, O595-596, 2014. (available here)
Contributing authors:
Edson Delatorre, Daiana Mir and Gonzalo Bello from Instituto Oswaldo Cruz, Fiocruz, Rio de Janeiro, Brazil.
These results are now published in MEM INST OSWALDO CRUZ, RIO DE JANEIRO, 112(11) NOVEMBER 2017. DOI: 10.1590/0074-02760170299 (available here)