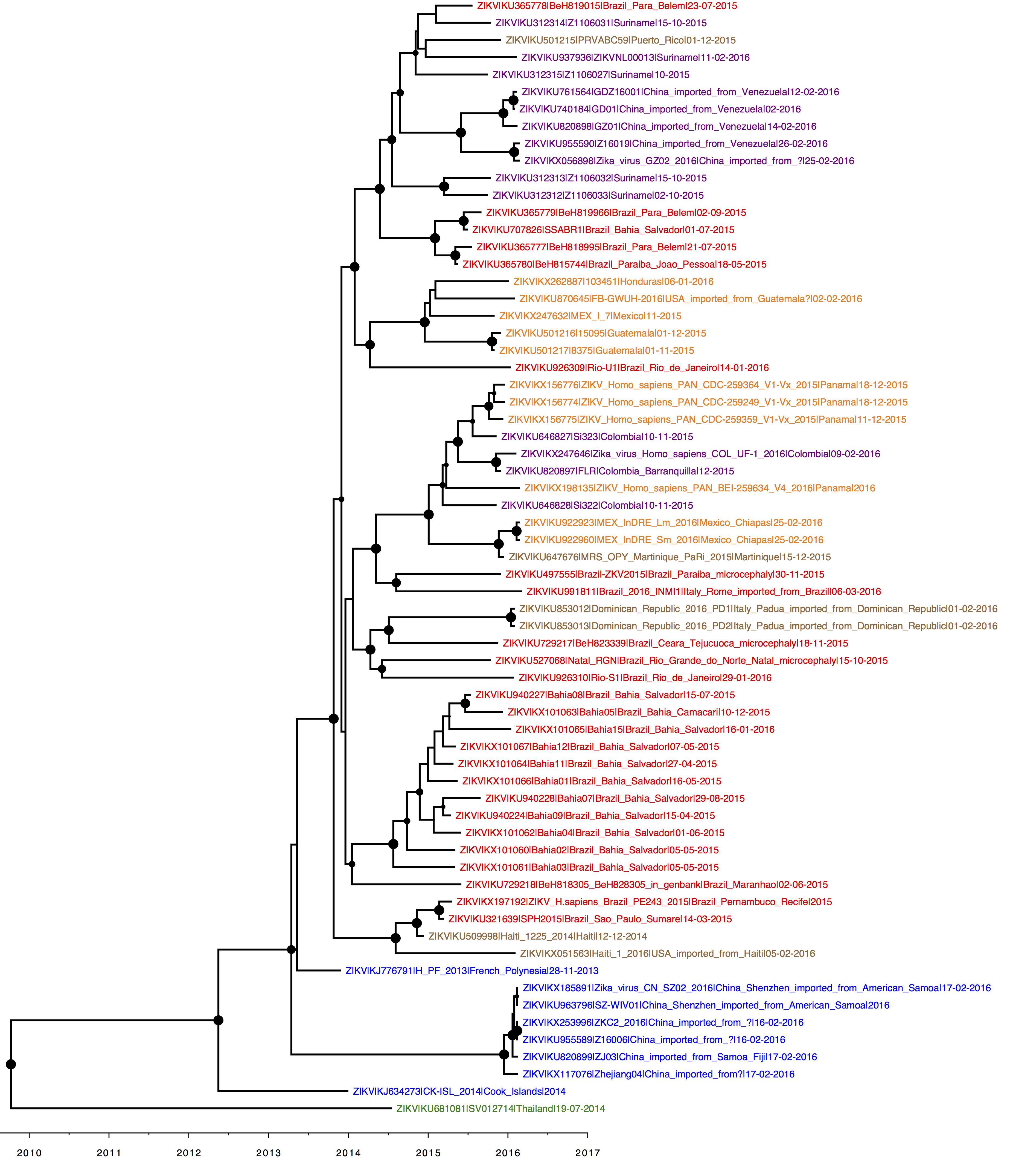
An updated clock tree, coloured by parsimony. Credit for the analysis to Julian Theze, credit for the sequences to the original data generators (see individual GenBank entries).
Brazil = red, Pacific = blue, central America = orange, south America excl. Brazil = purple, Caribbean = brown.
Fairly self-explanatory. Geographic structure becoming more apparent as number of isolates increase. Within the American lineage, Brazil isolates are commonly basal to non-Brazilian ones, with the exception of the Haiti strains. The large Salvador/Bahia clade is described here: http://biorxiv.org/content/early/2016/04/24/049916
Good luck to the Zibra team, looking forward to seeing the results.
update.pdf (30.4 KB)
Right, no pressure!
Check out http://zibraproject.github.io to see the plans!
Great, thanks for sharing Oli. How’s the clock on that tree - apparent at all?
Clock signal comes from older outgroup sequences that are not shown in the figure (as per our previous analyses).
Thanks Oliver - makes sense. I was just wondering if there would be any discernible signal within the current epidemic similar to e.g. Ebola, or whether historical samples (of which we have very few, with most likely being tissue culture adapted, as per our earlier discussion here on Virological.org) would be required to get a robust signal (like your earlier analyses/Science paper).
On that note, it would be super helpful if strains could be denoted as being passaged or not, so they can be excluded/highlighted on root-to-tips etc.
Yeah, totally agree - @kuhnjens has been pushing this hard and NCBI allows for this designation, but the implementation is definitely sub-optimal.
I think it’s pretty safe to say that >90% of the data out there has been through T/C - whether from historic samples or the current epidemic.