Thanks for the feedback Andrew and Richard. I’ve updated https://nextstrain.org/groups/blab/sars-like-cov to split ORF1a and ORF1b. This makes it clearer how nucleotide mutations map to amino acid substitutions.
Ignoring the divergent BetaCoV/Wuhan/IVDC-HB-05/2019 sequence and masking the initial 11 bases of the alignment, we have the following 5 strains and their mutations relative to the base of the outbreak clade:
WIV04/2019 - no mutations
IVDC-HB-01/2019 - no mutations
IPBCAMS-WH-01/2019 - 3 nucleotide mutations / 2 AA changes
IVDC-HB-04/2020 - 3 nucleotide mutations / 3 AA changes (includes C1023T and C1025T which are suspect being so close together)
The single basepair gaps are in homopolymeric runs suggesting a sequencing platform that maybe has problems with those. There are 2 larger deletions which I assume are missing read coverage.
I get slightly different stats on the # mutations - HB-04 has some indels that need corrections. Keeping HB-01 as the reference (should maybe be WH-01 though, as that’s the oldest sequence):
IVDC-HB-01/2019: [ref]
IPBCAMS-WH-01/2019: 3 mutations (2 non-syn / 1 syn)
WIV04/2019: 0 mutations
Hu-1/2019: 1 mutation (1 non-syn)
IVDC-HB-04/2020: 2 mutations (2 non-syn) (however, I don’t believe these, so I think this should also be 0 mutations)
I agree with Trevor that the mutations in HB-04 are suspect - right next to each other, non-synonymous, close to a poly-T stretch, and this sequence also needed some manual editing for indels. I think these are probably not correct and that sequence would then also be identical.
As for IVDC-HB-05, I agree with everybody that this sequence is definitely wrong (clustering of mutations, wacky ts/tv ratio, etc). If I do my very best to eliminate sequencing errors that I have commonly observed over the years, then I get a maximum of 7 mutations in this sequence, 4 of which are non-synonymous. These 7 can’t be excluded as likely errors (unlike the other 46 mutations in this sequence), but I think they still represent a (substantial) over-estimation.
Taking what I said above at face value and making WH-01 the reference (being the earliest sequence), we have three substitutions early in the tree (2 non-synonymous, 1 synonymous), followed by one additional substitution in Hu-1 (non-synonymous).
Thanks for checking this Kristian. I agree with you that the SNP at the end of Hu-1/2019 is spurious as is the SNP at 18460 in BetaCoV/Wuhan/IVDC-HB-04/2020 that appears directly adjacent to a long stretch of ambiguous bases. I’ve masked site 18460 as well as both ends of the alignment. With these changes our counts agree. Additionally, I thought it prudent to drop IVDC-HB-05 from the tree as its divergence is almost certainly spurious.
I think it would be unlikely that you would get zero coverage just for 1 basepair. The fact that they are in homopolymeric runs suggests systematic run-length errors. This is probably not Illumina data.
Several sequences including Thailand cases has been added to GISAID today.
Although GISAID announced their whole genome analysis result, I am wondering if you would update your analysis, since your analysis provide much more information including diversities.
Sincerely.
The Zhejiang Provincial Center for Disease Control and Prevention has shared two new genomes via gisaid.org. We’ve updated https://nextstrain.org/ncov to include them in our analysis bringing total up to 15 highly related samples.
Four more genomes were released, bringing the total to 19. Note that a couple of these look suspicious:
EPI_ISL_403928: A lot of mutations - can’t be trusted at this stage
EPI_ISL_403931: Mutations in the 5’ end that are wrong
Still not a lot of diversity.
Based on this dataset I count 17 SNPs that appear to be real and 35 that do not (this is not including indels in 402120). All SNPs are private - none of them transmitted.
Thanks @Kristian_Andersen. We’ve updated https://nextstrain.org/ncov accordingly, but have also left out Wuhan/IPBCAMS-WH-05/2020 / EPI_ISL_403928 due to appearance of spurious mutations.
However, I’d note that if there’s been multiple spillover events from the animal reservoir, I would really expect to be seeing clusters of genetically distinct human cases. So, a divergent sequence by itself is an expected thing. What threw me here however, was strange clustering of mutations in Wuhan/IPBCAMS-WH-05/2020.
Yeah, we can’t a priori assume that a divergent sequence is wrong, but for all sequences in this set where there were multiple mutations, many (all) of them were clearly the result of sequencing errors. Even after taking out the most ‘offensive’ sequences, I’m still fairly certain ~50% of SNPs are sequencing errors.
Doing some quick calculations on a SARS alignment from the 2002 epidemic, I get an N/S rate of ~0.5 and a Ts/Tv of ~ 3.5 - meaning that while synonymous and/or transitions are more frequent, but maybe not as much as we would typically see with other RNA viruses (e.g., Ts/Tv typically ~8). Just as a reference for trying to weed out sequencing errors and bad sequences.
https://nextstrain.org/ncov has been updated with 6 new genomes sampled from Guangdong shared by the Guangdong Provincial Center for Diseases Control and 3 new genomes sampled from Wuhan shared by Institute of Pathogen Biology, Chinese Academy of Medical Sciences.
We now observe clustering of related infections. These are a cluster of two infections in Zhuhai (Guangdong/20SF028/2020 and Guangdong/20SF040/2020) and a cluster of three infections in Shenzhen (Guangdong/20SF013/2020, Guangdong/20SF025/2020, Guangdong/20SF012/2020). These are noted in GISAID as “family cluster infection” and reflect two separate family clusters (https://twitter.com/JingLu_LuJing/status/1220143773532880896). This almost certainly represents human-to-human transmission.
Hopefully this isn’t too off-topic but I thought I’d put it here in case others are also interested in this application of sequences - we are looking into starting animal model work and are not sure what viruses we could get or whether we’d need to try and make our own via reverse genetics from synthetic gene constructs. Based on what we know now, is there a sense of which sequence or sequences would be best to use?
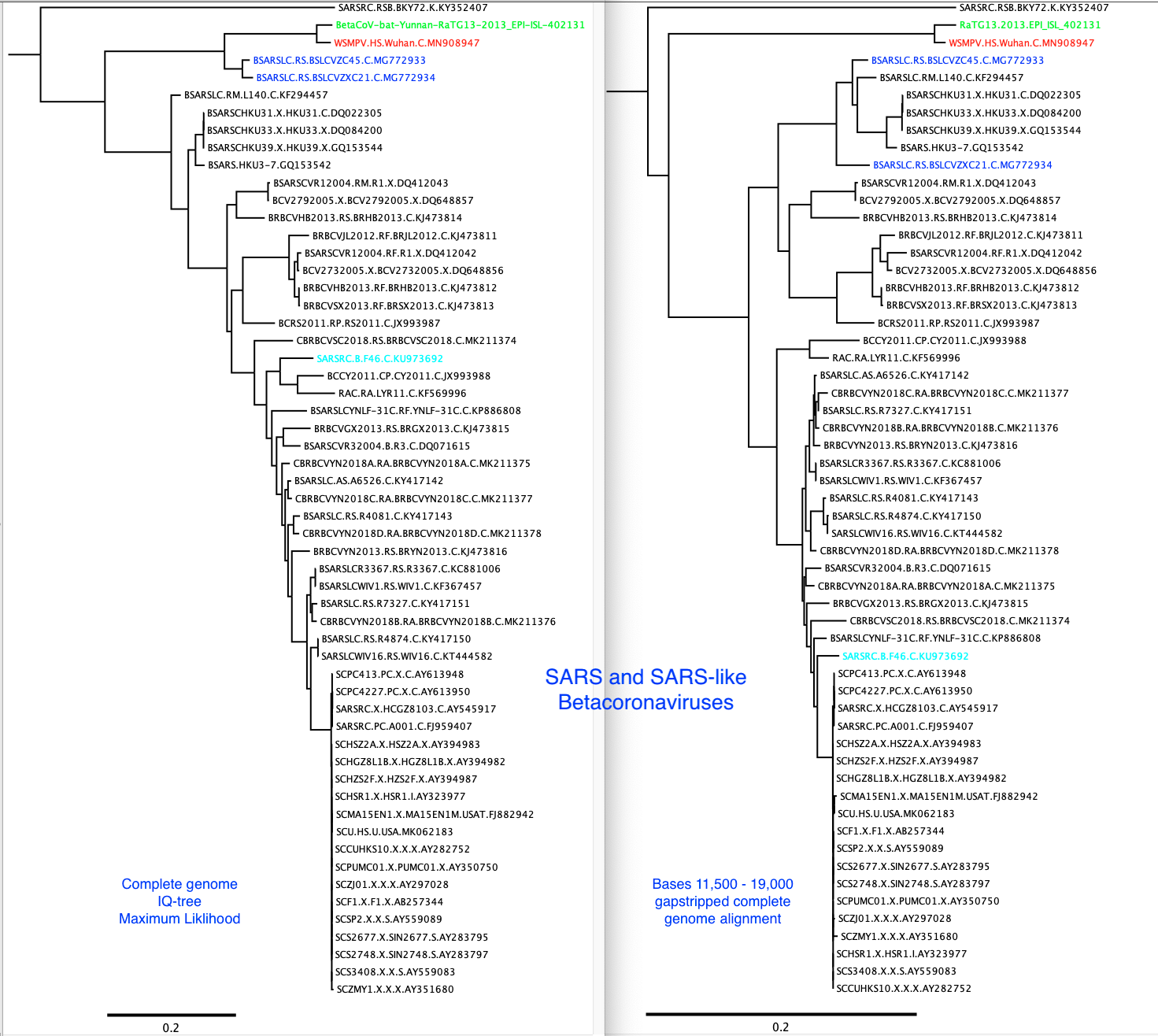
A 2013 bat Betacoronavirus (GISAID accession EPI_ISL_402131) was recently uploaded to GISAID. It is much more closely related to the 2019-2020 human outbreak viruses than the previously available bat virus genomes.
The publication of this sequence is available at the BioRxiv:
There is recombination between many of these viruses, so the phylogenetic trees built from complete genomes is not an accurate reflection of the evolutionary history of the various lineages.
A coronavirus was recently found in short read sequence data from a pangolin viromics data set. I am attaching an alignment of the complete genomes, a maximum likelihood tree built from just the spike glycoprotein gene, that does not include the pangolin sequence and a tree built from complete genomes that does include the pangolin sequence. The 2013 Yunnan bat virus sequence is still closer to nCoV than the pangolin virus sequence, but the pangolin virus is quite close. The 2013 Yunnan bat virus is removed from the alignment uploaded here, because it is GISAID data.
The 2013 Yunnan bat Coronavirus genome sequence is now available from GenBank, with accession number MN996532. So I am now uploading an alignment with this sequence included.