I have put together a figure of the recombination pattern of some of the closest viruses to SARS-CoV-2 including RaTG13 and the pangolins. Includes the region indexed as 1680-3014 by David, above, although I call the breakpoints as 1455-2836. The entire region after the spike is just lumped together.

Figure 1 | Phylogenetic trees for regions across the genome of SARS-CoV-2 and related betacoronaviruses.
spike protein
Here is a similar diagram for the spike protein. Generally the bat virus, RaTG13, is the closest to the SARS-CoV-2 virus across the whole spike with the exception of the small variable loop region in the C-terminus domain (the receptor binding domain). In this region, RaTG13 suddenly leaps away in divergence leaving the pangolin virus, Guangdong/1/2020 as the closest. This suggests that RaTG13 acquired this different loop region by recombination with another bat virus.
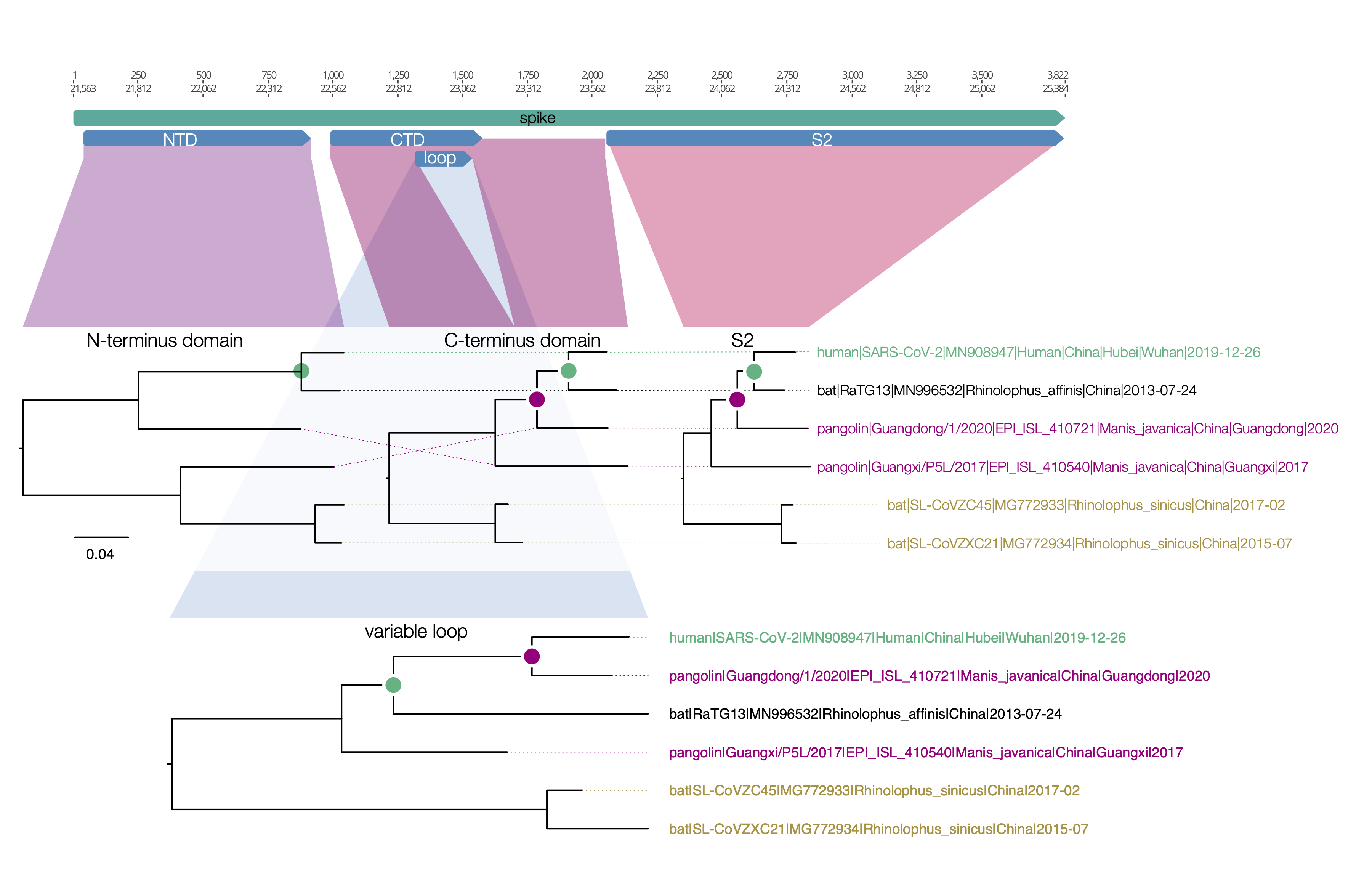
Figure 1 | Regions of the spike protein SARS-CoV-2 and its closest relatives. Trees are drawn to the same scale.
What is interesting about this loop region is that it contains the six key contact residues for the ACE2 receptor (see this post for more details about the RBD). So this suggests the common ancestor of the RaTG13 virus, the pangolin and the SARS-CoV-2 had the optimal receptor binding domain for ACE2 and then RaTG13 lost it.