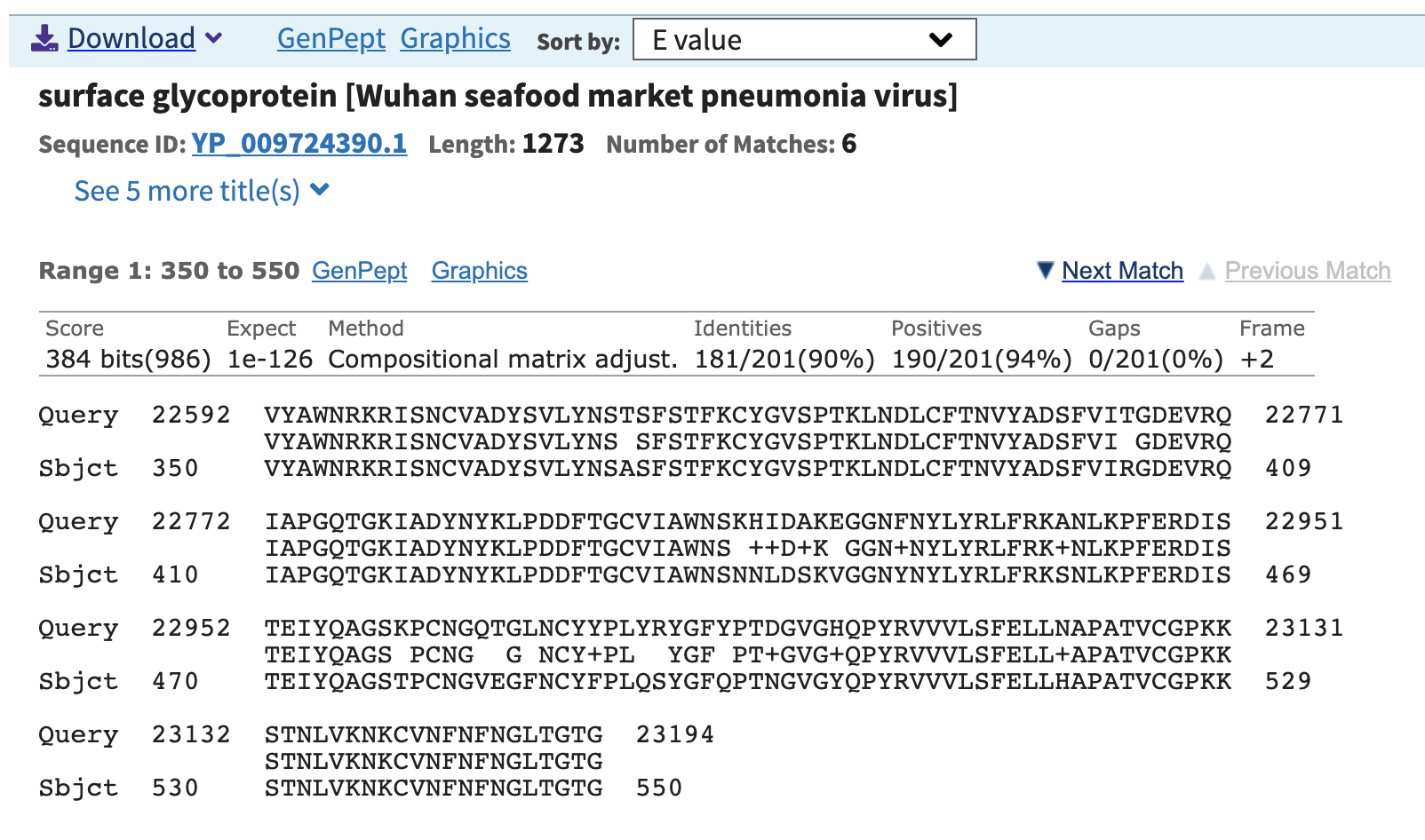
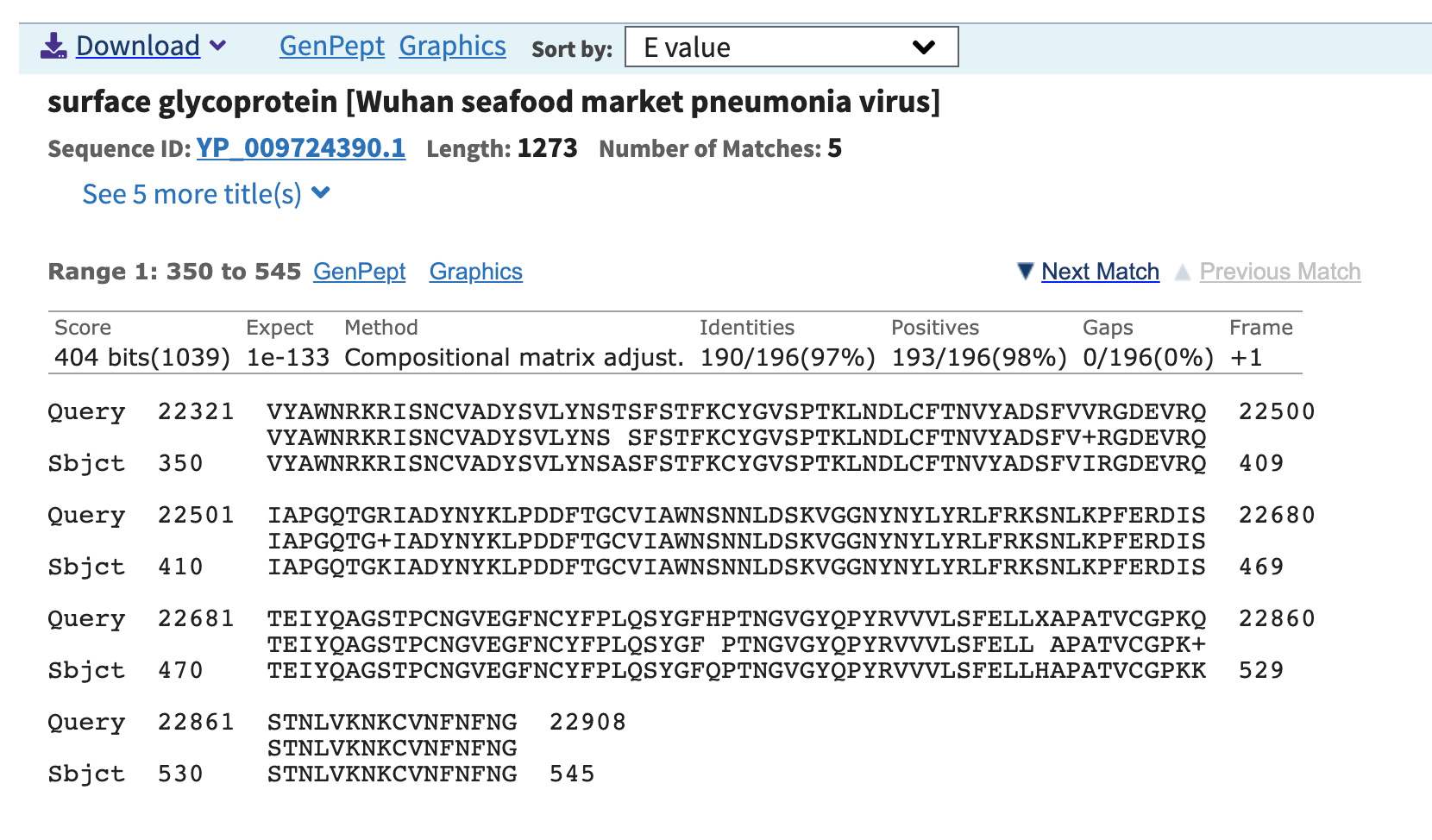
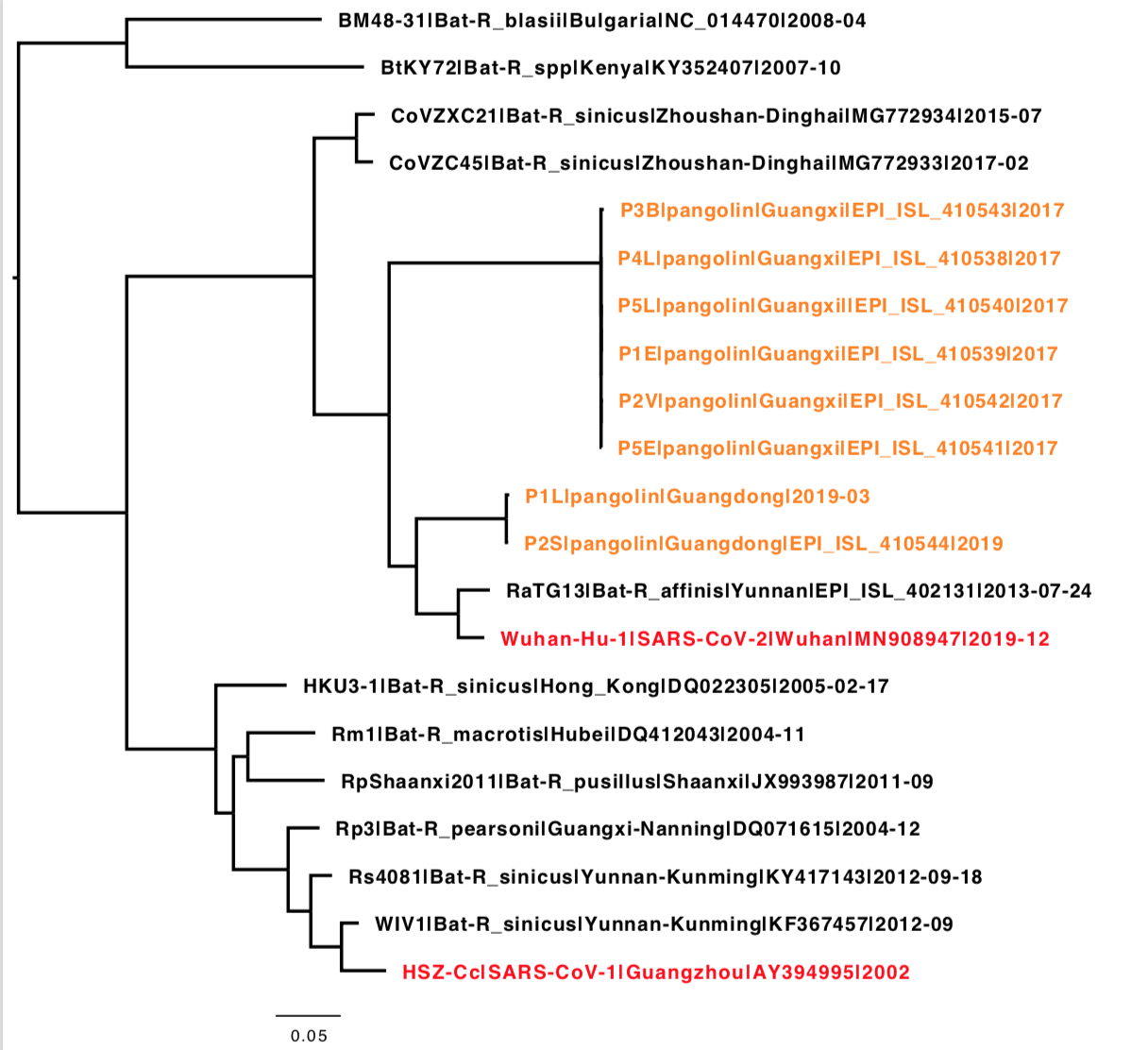
An outbreak of respiratory illness caused by a novel coronavirus (nCoV-2019, NC_045512.2) first identified in Wuhan China has resulted in over seven thousand confirmed cases. So far, the nCoV-2019 has been reported to share 96% sequence identity to the RaTG13 genome (EPI_ISL_402131). However, the S1 Receptor Binding Domain (RBD) of the nCoV-2019 genome was noticeably divergent between the two at amino acid residues 350 to 550 – Figure 1A. We aimed to identity coronaviruses related to nCoV-2019 in viral metagenomics datasets available in the public domain. In a recently published dataset describing viral diversity in Malayan pangolins (PRJNA573298) we used VirMAP to reconstruct a coronavirus genome (approximately 84% complete from samples SRR10168377 and SRR10168378) that shared 97% amino acid identity across the same RBD segment – Figure 1B. This result indicates a potential recombination event for nCoV-2019.
Edit -
From the coordinates shown in this preprint (Figure 4), it looks like most of the differences between RaTG13 and nCoV-2019 are restricted to loop 2 of the receptor binding motif (positions ~450-500).
The virological site does not allow me to upload a FASTA format multiple sequence alignment. But I have one and anyone who wants a copy can send me an email.
The complete genomes codon-aligned have a few small regions in individual sequences which are not “optimally” aligned. But overall, having a DNA alignment that translates to amino acids in one frame, is useful for studying selection pressure and other things.
The spike protein from the Wuhan strain is closer to RaTG13 overall, probably due to the S1-NTD subdomain being so different in the pangolin coronavirus. From the S1-CTD section on, the Wuhan strain and the pangolin strain are pretty similar (~97%), except for the furin cleavage site insertion.
It seems unlikely that the receptor binding domain–and especially the receptor binding motif–would be nearly identical to one found in pangolin through random chance.
Many news outlets now reporting pangolins as potential source. Saw it first on Reuters citing work from South China Agricultural University and claiming 99% genome similarity. But have not seen a preprint or the genome. Has anyone been able to track this?
which links to the same paper, and short read data, that torptube already posted. So apparently they are planning to publish what Matthew Wong already put up here. But maybe they will have a slightly different assembly.
This is new data that’s not been released yet. Xiaowei Jiang @john_jxw posted a translation on Twitter from the South China Agricultural University press release: “The team analyzed more than 1,000 metagenomic samples and identified pangolins as potential intermediate hosts of the new coronavirus; then, through molecular biological testing, it revealed that the positive rate of beta coronavirus in pangolins was 70%. … By analyzing the genome of the virus, they found the sequence similarity between the isolated virus strain and the currently infected human strain is as high as 99%.”
The title of this post ideally needs updated as Matthew Wong’s reconstructed virus from the dead pangolin metagenomic data from Liu et al 2019 (https://www.mdpi.com/1999-4915/11/11/979/htm) is relatively close to the 2019-nCoV lineage throughout its genome, not just in Spike. It’s not, however, closer than the RaTG13 genome from bats.
Thank you, Brian! I saw that but was left with curiosity to see the data. Matt and I worked on this together with a couple of more people from Baylor College of Medicine. The analyses were all based on available datasets and wished we would have found more datasets from pangolins. Maybe the SCAU investigators have more data. Let’s see!
My post from 6 days ago has my alignment of the pangolin virus genome to over 100 other Sarbecovirus genomes (SASR_SARSlikePlusPangolinCodonAligned.FASTA.gz above). Sorry for the typos and funny file name, it was supposed to be SARS and SARS-like viruses. Anyway, I have tweaked that alignment a bit more in the past 6 days, so if you want a very slightly improved version, write to me. I also have similarity plots and trees and other stuff like that.
You can see the bit in RaTG13 in the region by the key sites of the RBD (top row of each sequence is nucleotides, bottom amino acids). Also there is re recombinant region in the pangolin at the 5’ end of spike (it doesn’t extend into ORF1b).
So my reading of this is that the common ancestor of RaTG13, the pangolin and the lineage that leads to SARS-CoV-2 might have had those RBD residues but the bat got a recombination in and lost them.
I think it is important to keep in mind that the distances and thus the inferred dates of the common ancestors of these “closely related” viruses (RaTG2013 Yunnan Bat, and the pangolin viruses) are quite a way back in the past, as has been discussed on another thread here . These viruses are known to recombine, but I don’t think we have a good idea yet what type of timescale is involved. Do they recombine on average once every 40 years, or once every 400 years? The BioRxiv paper by Lam et al has trees built from subgenomic regions indicating that in many parts of the genome there seem to be two major clades. But recombination between those clades has happened, as is illustrated by the similarity plot of the YN2018A.MK211375 virus attached here.
In MERS the evidence is that recombination is frequent, often involves relatively small tracts, and occurs over the course of the few years we have been observing it in camels: https://academic.oup.com/ve/article/2/1/vev023/1753363