Follow-up on the flawed logic of codon bias linking snakes to 2019-nCoV
Along a similar vein to what Kristian Anderson’s group pointed out, we conducted related analyses that illustrate that codon bias analyses do not provide evidence that snakes were hosts of the 2019-nCoV virus. The conclusion by Ji and colleagues that snake were the most plausible reservoir for the 2019-nCov was based on their finding that snake genomes show greater codon bias to this virus than do genomes from bats or other plausible hosts, which as many of you on this forum have already pointed out is not a robust way to infer the host reservoir of a virus. By analyzing their data and additional genomic codon bias from other eukaryotes and coronaviruses, we further emphasize why this is a flawed inference.
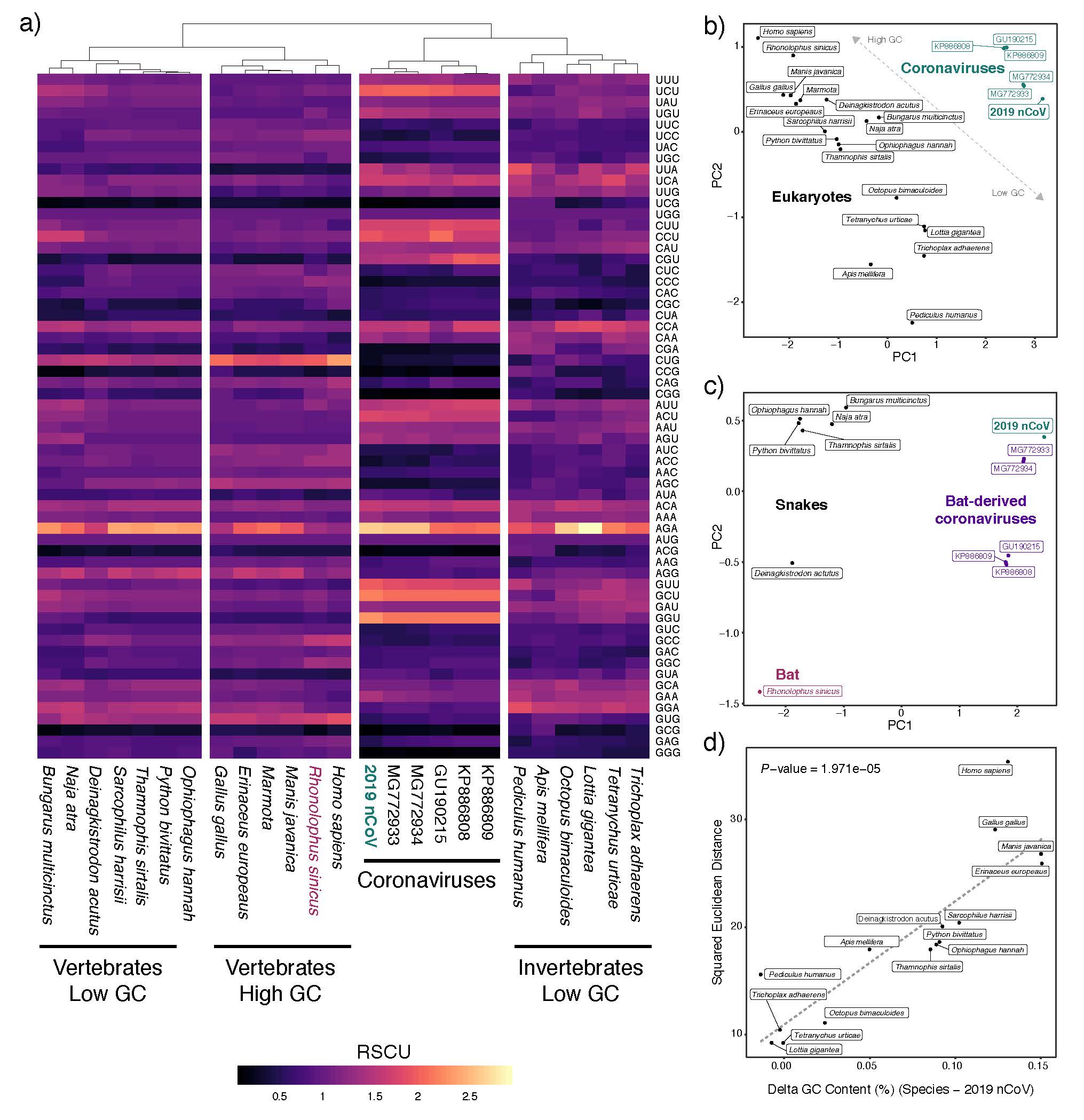
We show that the inherent AT-bias of coronavirus genomes in general leads to spurious inferences linking snakes (which also exhibit particularly AT-rich genomes) as hosts of 2019-nCoV . We conducted expanded analyses of codon biases from multiple coronaviruses isolated from bats with 2019-nCoV, and multiple additional snakes and other eukaryotes. These analyses underscore the lack of evidence from codon bias analyses for snakes as a host of 20190nCoV:
Synonymous codon usage patterns in genomes of coronaviruses isolated from bat hosts show nearly identical patterns as that of 2019-nCoV, indicating that, even if codon usage similarity was a valid method to determine the likely host of the virus, there is no evidence for 2019-nCoV having a divergent pattern of codon usage from viruses isolated from bat hosts (panel A). PCA analyses of codon biases among eukaryotes and coronaviruses further illustrate that codon bias alone is uninformative for linking eukaryotic hosts to particular viruses (panel B and C), and fails to implicate snakes being any more similar to 2019-nCoV than to other coronaviruses which were isolated from bats (panel C).
Coronaviruses tend to have relatively AT-rich genomes which is linked to their highly AT-rich codon biases. Accordingly, there is a linear relationship in which other AT-rich eukaryote genomes exhibit more similar codon usage to the AT-rich coronaviruses than do more GC-rich species. Many of these highly AT-rich genomes are implausible eukaryote hosts, but included to illustrate the point that codon bias simply links coronaviruses with highly AT-rich eukaryote genomes. As snakes are particularly AT-rich compared to other vertebrates, they inherently exhibited more similar codon usage to 2019-nCoV than did more GC-rich genomes of mammals, which led to Ji et al.’s incorrect conclusion about snakes.
Lastly, the snake species implicated by Ji et al. are also unlikely to naturally prey on bats. Both snake species are classical dietary specialists that prey almost exclusively on other snakes, and not on bats (as implied by Ji et al.).
-Todd Castoe & Blair Perry