First cases of coronavirus disease (COVID-19) in Brazil, South America (2 genomes, 3rd March 2020)
We provide a brief report and phylogenetic analysis of the confirmed COVID-2 cases in Brazil. From 488 suspected cases, two have so far tested positive for COVID-19. These two cases both travelled to Northern Italy. Detailed clinical and epidemiological descriptions for suspected and confirmed patients, including for the two patients reported here, are available from the National Public Health Emergency Alert and Response Network from the Brazilian Ministry of Health.
Case reports and diagnostic testing
Patient SPBR1 is a 61 year old male that travelled from São Paulo to Italy (Lombardia region) in early February 2020. Upon returning to São Paulo on 21st February, the patient presented with respiratory symptoms including fever, dry cough, sore throat, and runny nose. On 25th February 2020, the patient visited a hospital in São Paulo city and a nasopharyngeal swab was collected for virus detection. By 26th February 2020, confirmatory diagnostic real-time RT-PCR testing had been conducted at the Instituto Adolfo Lutz (IAL), the regional reference laboratory for virus detection in São Paulo state. The sample from patient SPBR1 had a RT-PCR cycle threshold (Ct) value of 30.
Patient SPBR2 is a 32 year old male that travelled from Italy (Milan city, Lombardia region) to São Paulo city on 27th February 2020. Respiratory symptoms including cough, sore throat, myalgia and headache started on the flight to São Paulo, and the patient wore a mask during the flight. On 28th February, the patient visited a hospital in São Paulo and a nasopharyngeal swab was collected. Virus infection was confirmed on the same day using real-time RT-PCR and viral RNA was sent IAL for genomic sequencing. Sample from patient SPBR2 had a RT-PCR Ct value of 18.
Genome sequencing of confirmed cases
To investigate the genomic diversity of these two cases, genomes were generated by the CADDE project at the IAL together with the Institute of Tropical Medicine team from the University of São Paulo (IMT-USP). We used the openly-available COVID-19 sequencing and bioinformatics protocols developed by the ARTIC network. Sequencing protocol, sequencing multiplex primers, and bioinformatic protocols are described in detail in Artic Network. cDNA synthesis was conducted in duplicate for each sample. The concentration of PCR products was measured using a Qubit dsDNA High Sensitivity kit on a Qubit 3.0 fluorometer (ThermoFisher).
Library preparation was conducted without a barcoding step and libraries were sequenced on an R9.4.1 flow cell. Sequencing was conducted in MinKNOW version 19.10.1 for over 12 hours. The open-source software RAMPART version 1.0.5 was used to assign and map reads in real-time. Raw files were basecalled with Guppy, demultiplexed and trimmed with Porechop and mapped against reference strain Wuhan-Hu-1 (GenBank accession number MN908947). Variants were called using nanopolish 0.11.3 and accepted if they had a log-likelihood score of greater than 200. Low coverage regions were masked with N characters. Example of read data for SPBR1 is available for inspection from https://cadde.s3.climb.ac.uk/covid-19/BR1.sorted.bam. Consensus genome sequences have been deposited in GISAID with the following IDs: SPBR1: EPI_ISL_412964 and SPBR2: EPI_ISL_413016.
Phylogenetic analysis of COVID-19 complete virus genomes
To put the Brazilian genomes into the context of the global epidemic, we added them to a dataset of 157 curated complete genomes available from GISAID, as of 3rd March 2020. A multiple sequence alignment of 159 complete genomes from 20 countries was generated using MAFFT and manually curated. A maximum likelihood (ML) tree was estimated using PhyML version 3.0 using a general time-reversible nucleotide substitution model with a proportion of invariant sites.
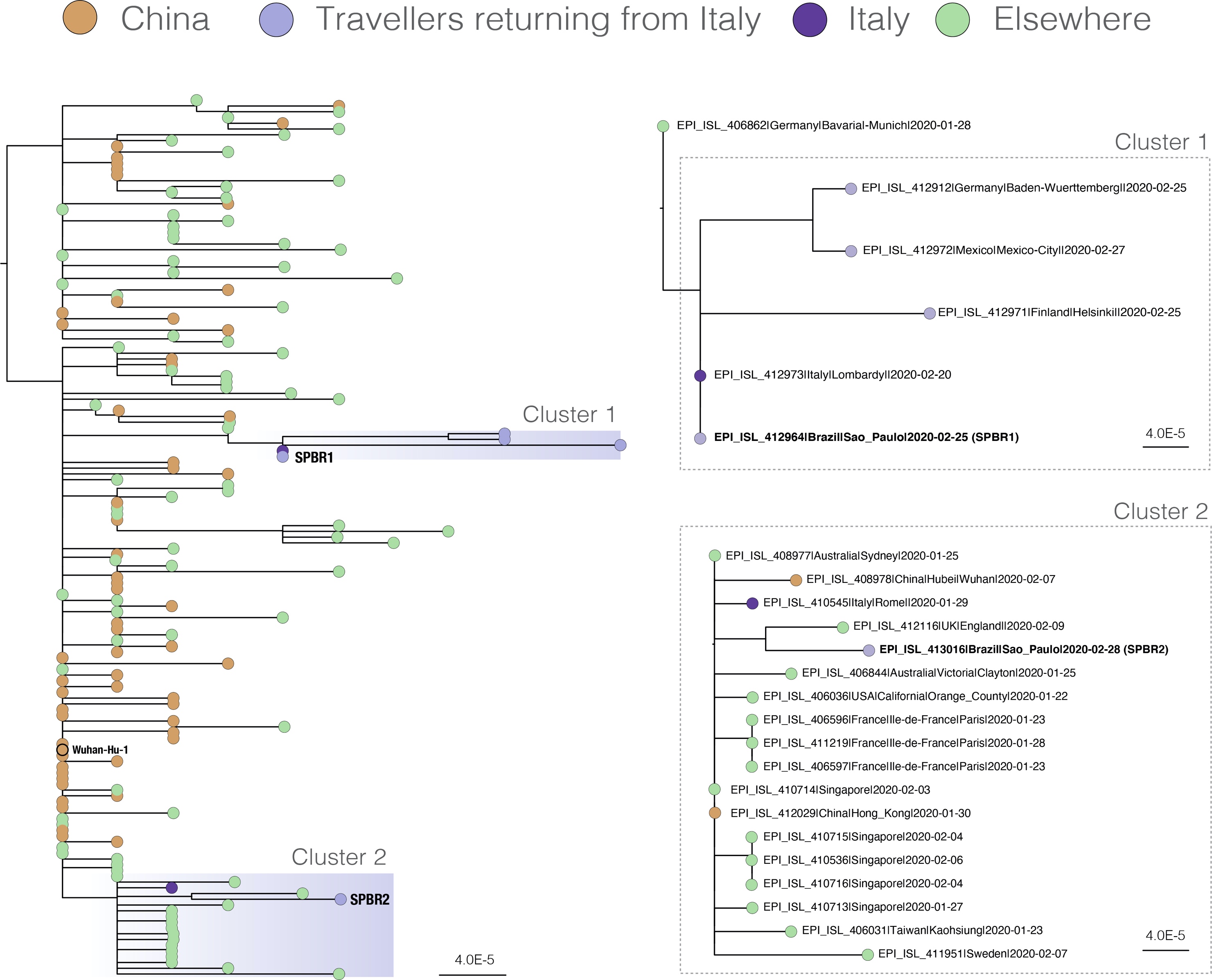
Figure 1. Estimated maximum likelihood phylogenetic tree for complete genome sequences from 159 COVID-19 virus strains. Genome data used here was kindly made available in GISAID, as 3rd March 2020). The right hand panels show zoomed views of Clusters 1 and 2 containing the Brazilian genomes.
Figure 1 shows the estimated maximum likelihood phylogeny. As expected, sequences from China are interspersed across the tree. Interestingly, the two infections samples in Brazil group in different phylogenetic clusters. SPBR1 groups in Cluster 1 and is identical to a recently released sequence from Lombardy, Italy (EPI_ISL_412973). Cluster 1 also contains sequences obtained from patients from Germany, Mexico and Finland that reported travelling to Italy. When combined with the patient travel information, this indicates that the two confirmed cases in Brazil are the result of separate introductions to the country.
The genome from patient SPBR2 groups in Cluster 2, which contains sequences from several countries including China, England, Australia, France, USA, Singapore, Taiwan, and Sweden. We note that the case from England (GISAID ID EPI_ISL_412116) represents a direct import to London from China (Figure 1). The SPBR2 genome sequence is separated by three mutations from EPI_ISL_408977, which represents a case acquired in China and sampled in Australia on 25th January, and by four mutations from sequence EPI_ISL_410545, obtained from a Chinese tourist in Rome, Italy sampled on 29th January 2020. Although it is possible that patient SPBR2 was infected by a traveller from another part of the world, the infection was more likely acquired in Lombardy given the current prevalence of COVID-19 circulating there. If that is true, then the outbreak in Northern Italy was likely the result of multiple introductions to the region and from not a single source.
This has implications for epidemiological tracing and for identifying connections to the Italian outbreak using genome sequence analysis. Incidentally, a few Swiss genomes have been deposited overnight which seems to confirm the hypothesis of multiple virus introductions to Northern Italy. We caution, however, that further analysis is needed to evaluate the statistical robustness of the clusters in the SARS-CoV-2 phylogeny, which is challenging at this stage of the epidemic due to the small number of mutations between sequenced strains.
In conclusion, genome sequencing and analysis of the first two COVID-19 virus genomes from Brazil shows two independent virus introductions to the country. Phylogenetic analysis is consistent with the reported travel history of SPBR1. The challenges in inferring directionality of transmission for SPBR2 based on genetic data alone can be explained by undersampling and limited genetic variation of currently available virus genomes. Additional genome data from Northern Italy, and from travellers to that region, will help to elucidate the patterns of transmission there. Finally, continued surveillance of new cases will be critical to anticipate virus importations to Brazil, understand transmission in different settings, and identify possible clusters of local transmission in the country.
Disclaimer
The new sequences have been deposited in GISAID with accession IDs EPI_ISL_412964 (SPBR1) and EPI_ISL_413016 (SPBR2). Please feel free to download, share, use, and analyze the data from Brazilian strains deposited in GISAID. We ask that you communicate with us if you wish to publish results that use this data in a journal. If you have any other questions, please also contact us directly.
Contributing authors
Jaqueline Goes de Jesus, Claudio Sacchi, Ingra Claro, Flávia Salles, Erika Manulli, Daniela da Silva, Terezinha Maria de Paiva, Margarete Pinho, Ana Maria Sardinha Afonso, Andressa Mathias, Lincoln Prado, Ana Lucia de Carvalho Avelino, Katia Correa de Oliveira Santos, Filipe Romero, Fabiana dos Santos, Claudia Gonçalves, Maria do Carmo Timenetsky, Joshua Quick, Oliver G. Pybus, Nick Loman, Andrew Rambaut, Ester C. Sabino, Nuno R. Faria
Affiliations
Laboratorio Estratégico, Instituto Adolfo Lutz, São Paulo, Brazil
Núcleo Doenças Respiratórias, Centro de Virologia, Instituto Adolfo Lutz, São Paulo, Brazil
Centro Nacional de Influenza, Instituto Adolfo Lutz, São Paulo, Brazil
Instituto Medicina Tropical, Universidade de São Paulo, Brazil
University of Birmingham, United Kingdom
Universidade Federal do Rio de Janeiro, Brazil
Institute of Evolutionary Biology, University of Edinburgh, United Kingdom
Department of Zoology, University of Oxford, United Kingdom
Funding
FAPESP Medical Research Council Brazil-UP CADDE partnership award (MR/S0195/1), a Wellcome Trust and Royal Society Sir Henry Dale Fellowship (204311/Z/16/Z), Oxford Martin School, and a Wellcome Trust Collaborators Award 206298/Z/17/Z (ARTIC network).
Acknowledgments
We would like to thank all the authors who have kindly deposited and shared genome data on GISAID. A table with genome sequence acknowledgments can be found heregisaid_cov2020_acknowledgement_table.xls.zip (28.5 KB).
Updated epidemiological situation in Brazil:
http://plataforma.saude.gov.br/novocoronavirus/
Notification of suspected and confirmed cases in Brazil:
https://redcap.saude.gov.br/surveys/?s=TPMRRNMJ3D
Additional information (in Portuguese):
Contact information
Professor Ester Cerdeira Sabino, MD, PhD
Instituto Medicina Tropical, University of Sao Paulo, Brazil
Email: [email protected]
Professor Nuno Rodrigues Faria, PhD
Associate Professor, University of Oxford, United Kingdom
Email: [email protected]