Following up on our first post on the sequencing efforts by the collaboration between the Irrua specialist Teaching Hospital (ISTH), Institute for Lassa Fever Research and Control (ILFRC), Irrua, Edo State, Nigeria, The Bernhard-Nocht Institute for Tropical Medicine (BNITM), Hamburg, Germany, Public Health England (PHE), we now make a available sequences from 8 additional 2018 samples, amounting to a total of 15 samples:
https://github.com/ISTH-BNITM-PHE/LASVsequencing
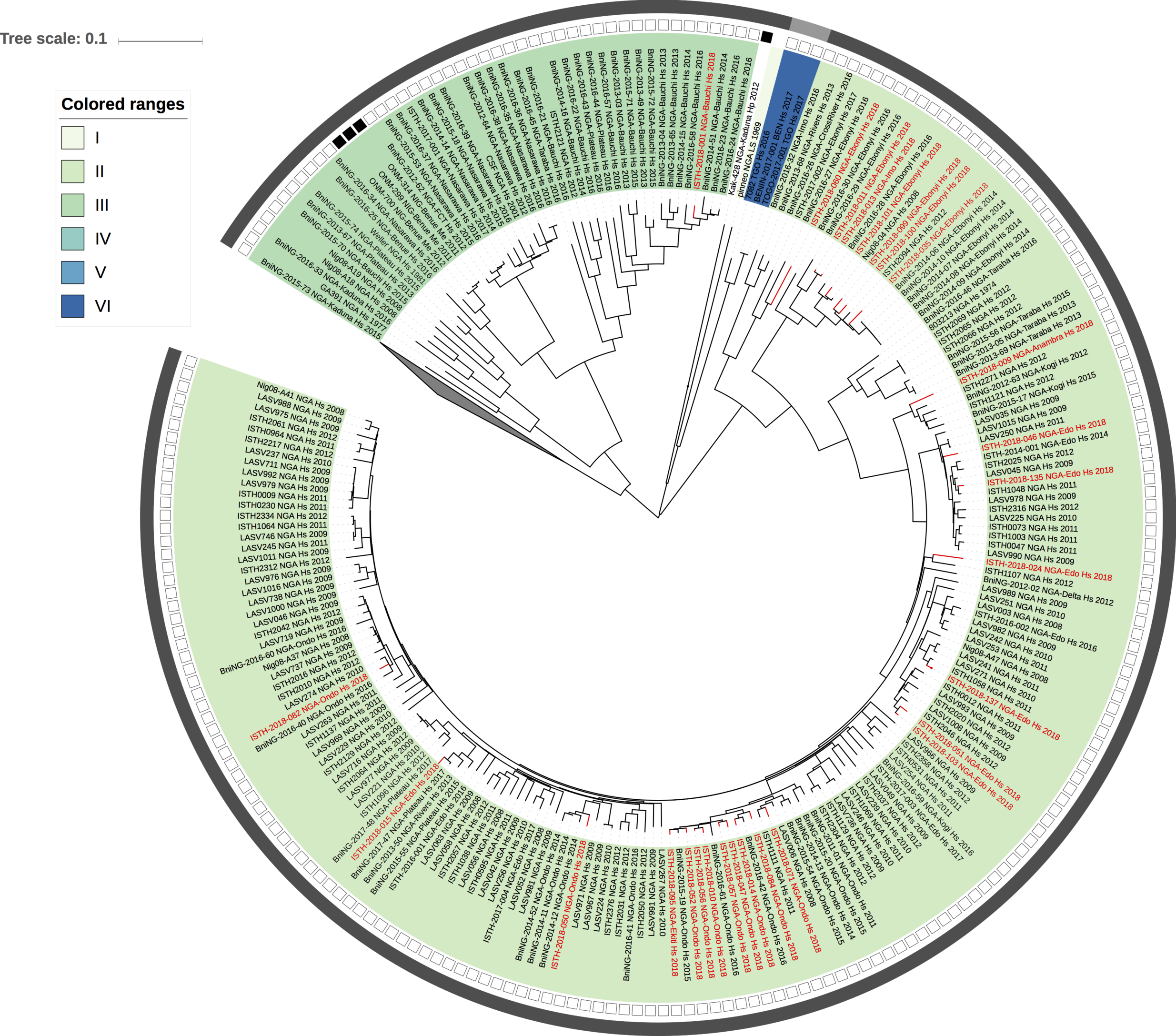
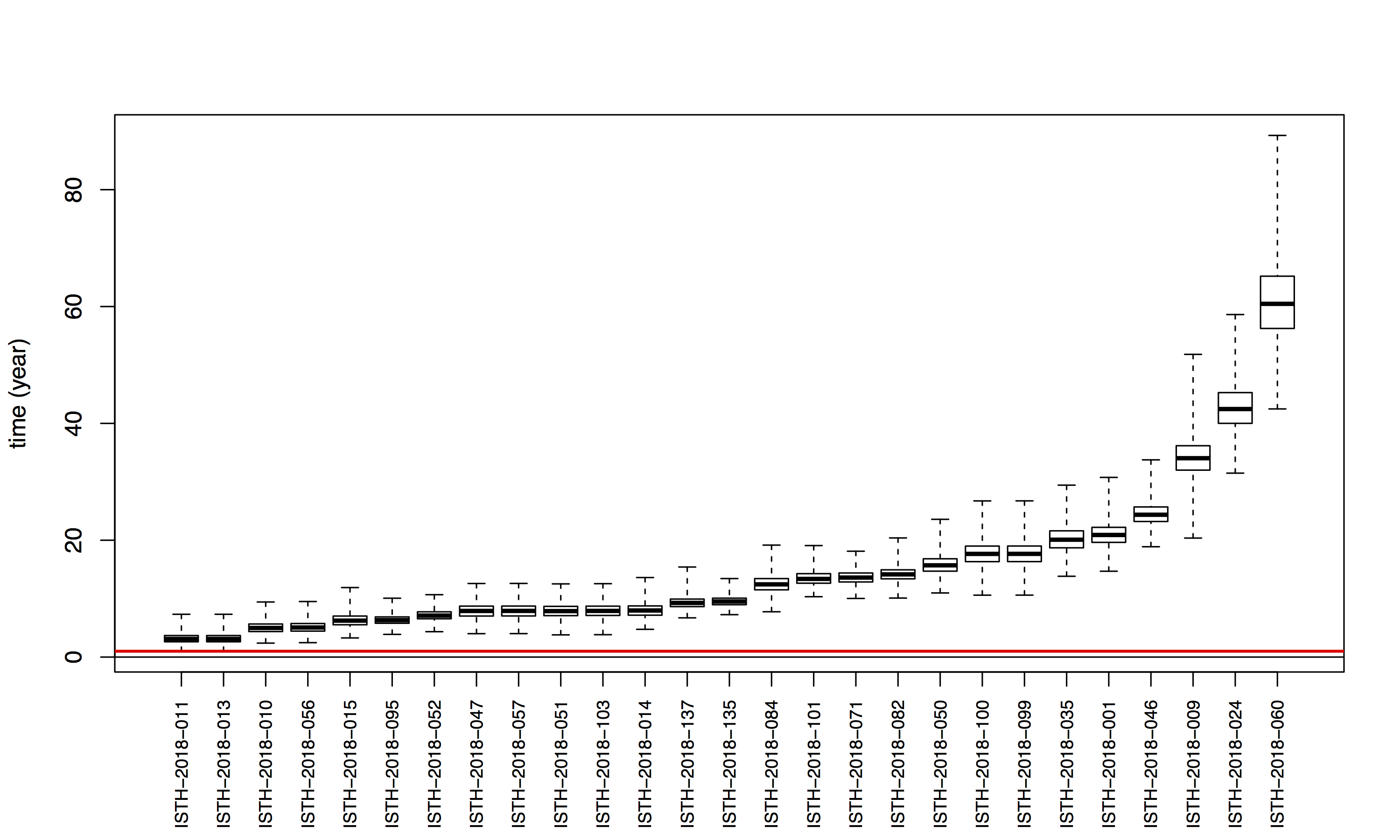
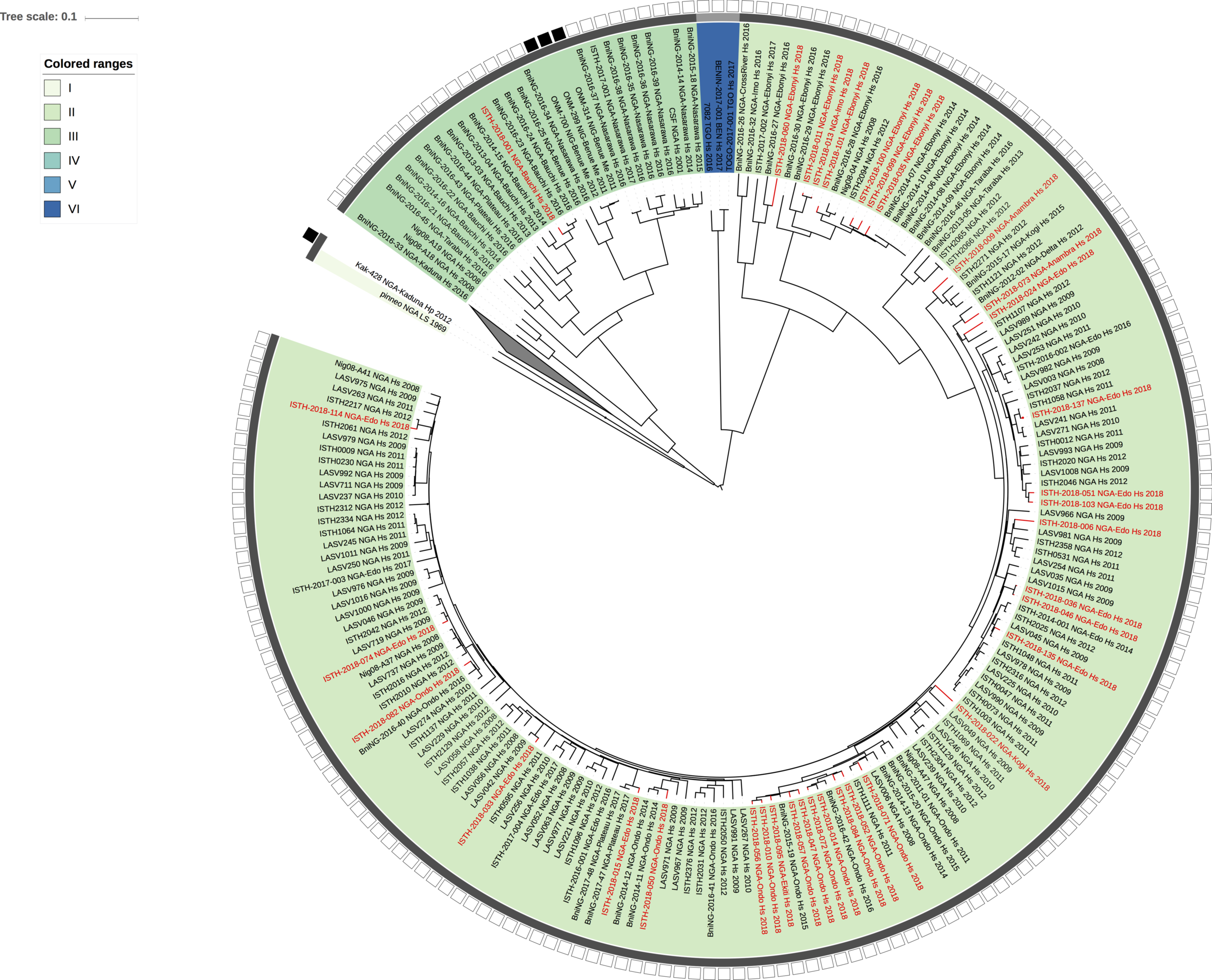
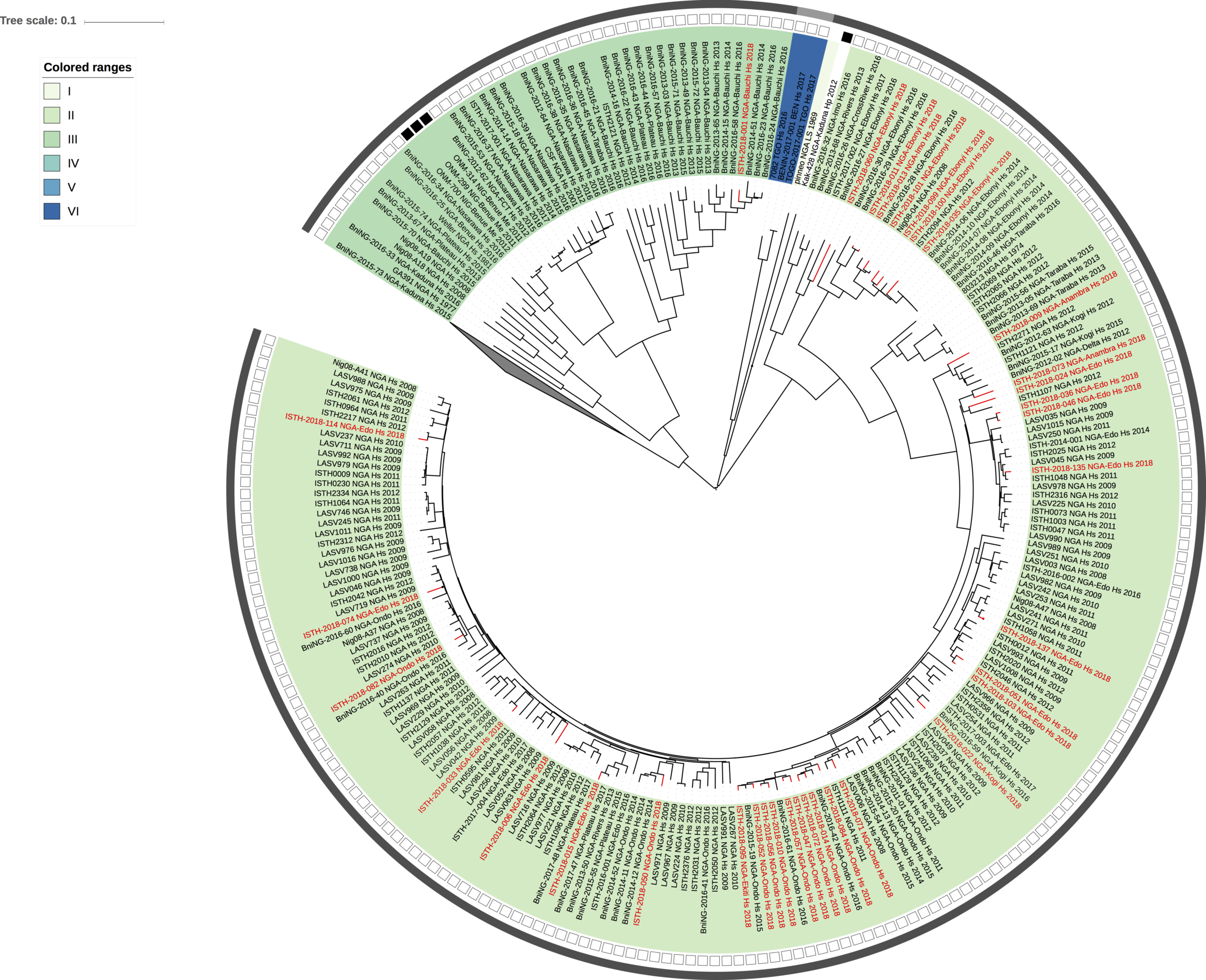
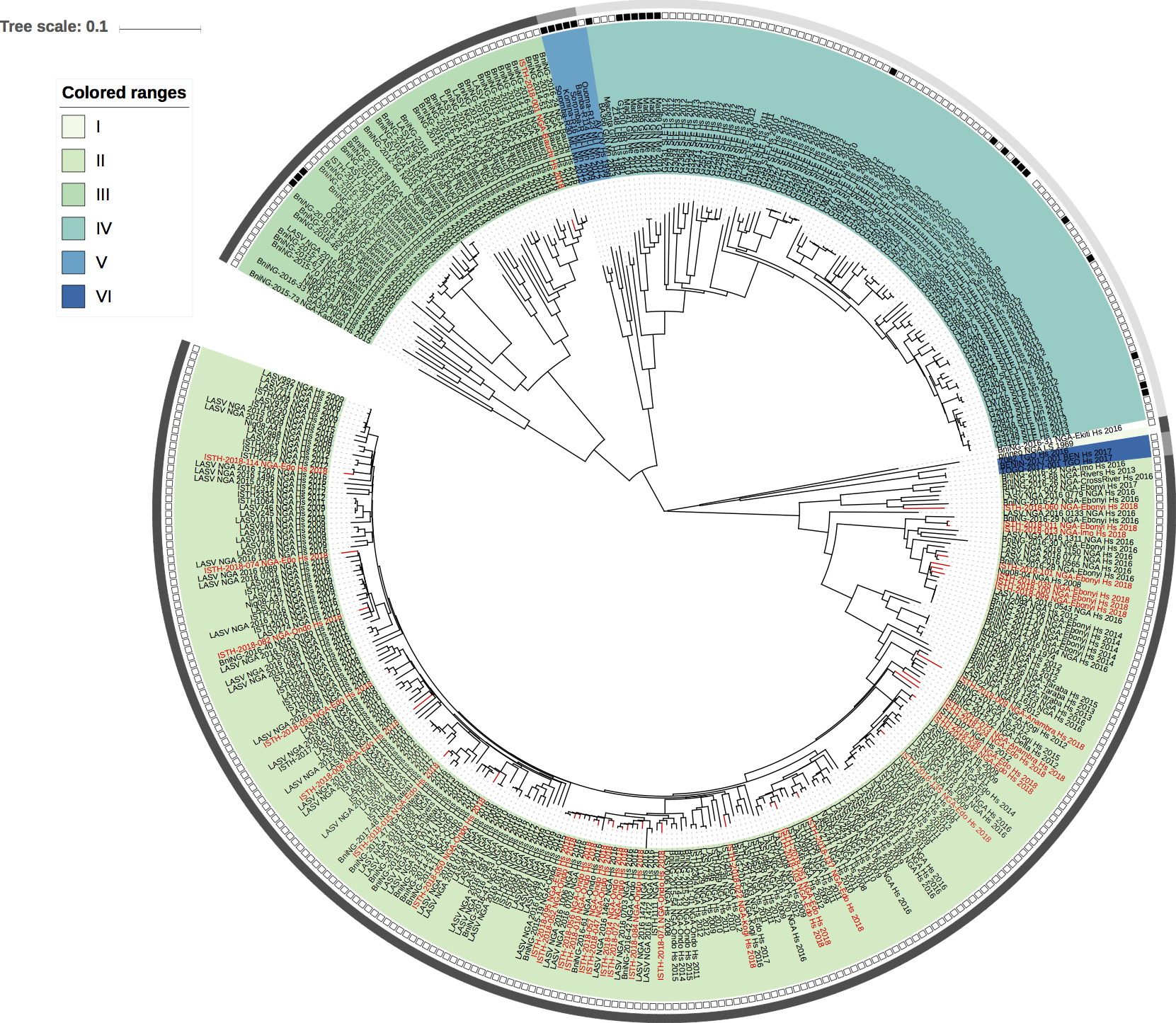
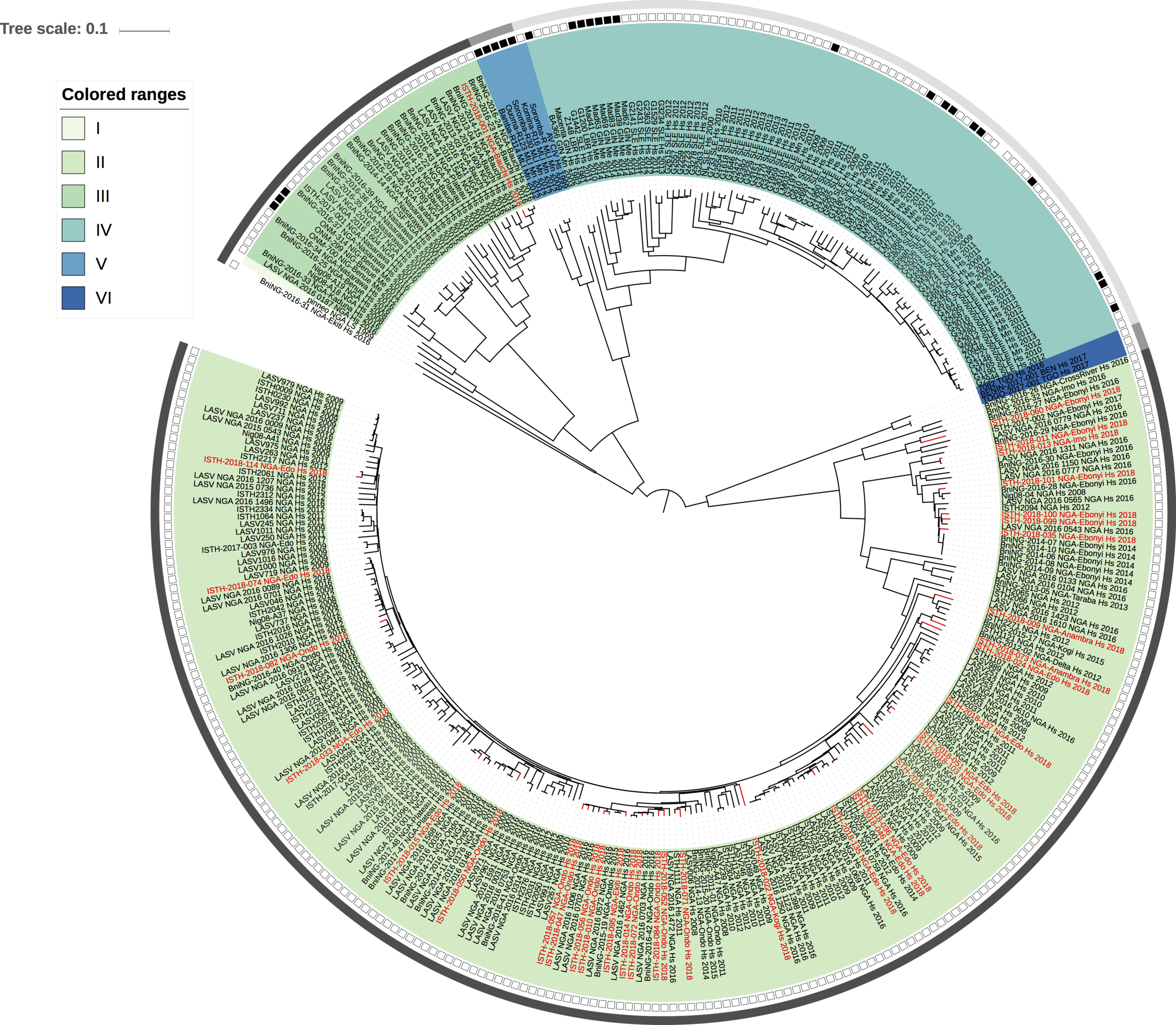
Maximum likelihood phylogenetic analyses of the coding genes in both the L and S segments indicates more spill-over from the rodent hosts, now also in lineage III. However, we start to observe clusters of pairs of 2018 viruses (ISTH-2018-010 & ISTH-2018-056, and ISTH-2018-011 & ISTH-2018-013). In order to estimate the time of their most recent common ancestors (MRCAs), we performed BEAST analysis on the relevant subset of the S segment data. Divergence times and evolutionary rates are consistent with those previously estimated by Andersen et al. (doi: 10.1016/j.cell.2015.07.020). The time estimates demonstrates that the MRCAs dates back to 2013 (2011-2015) for ISTH-2018-010 & ISTH-2018-056 and to 2015 (2013-2016) for ISTH-2018-011 & ISTH-2018-013. So, this makes human-to-human transmission also unlikely for these pairs, indicating that -as for the other viruses - they are the result from independent reservoir spill-over. As a more dense sampling will become available, we expect to see more clustering of 2018 strains.
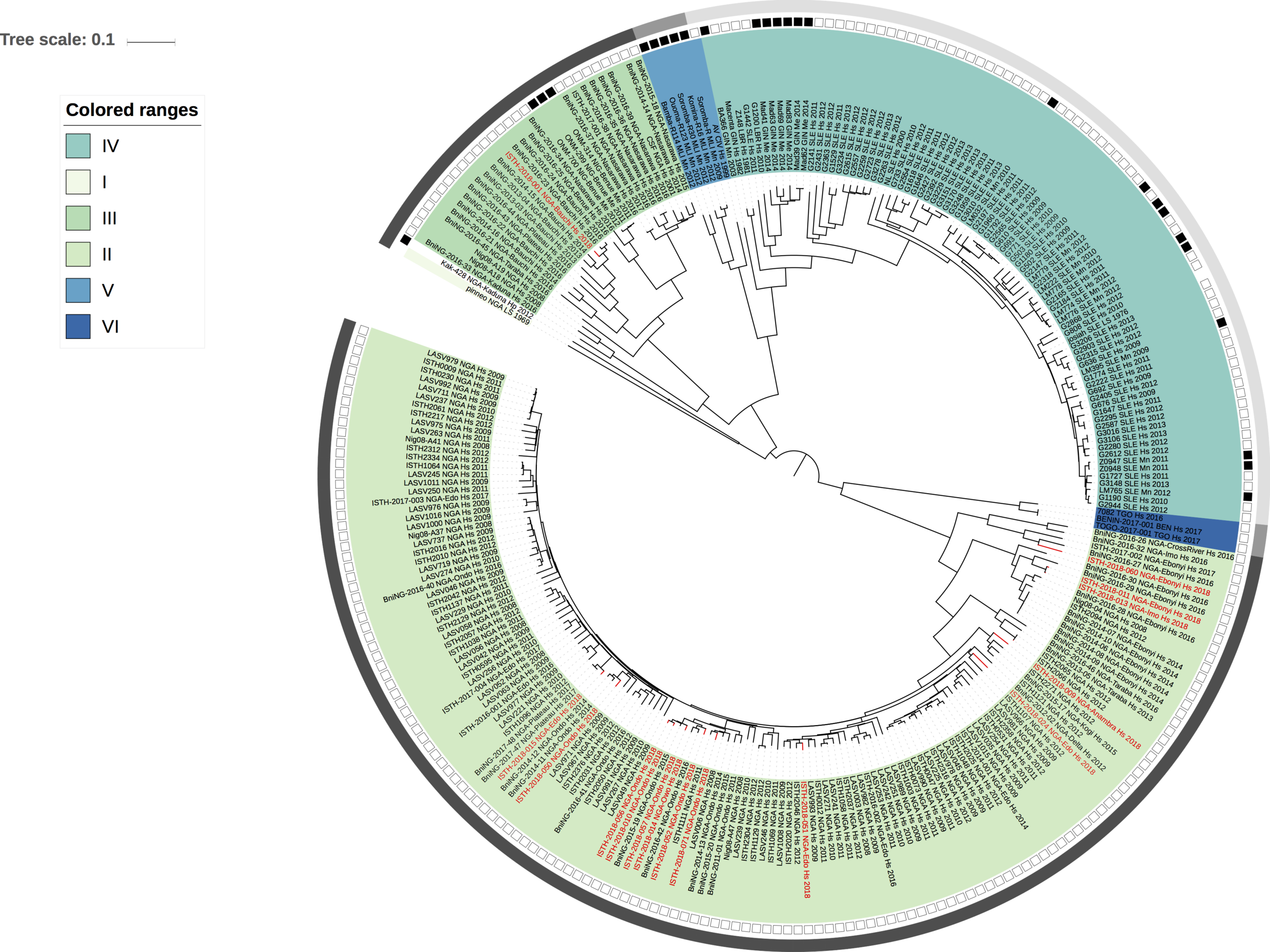
Figure 1: Lassa virus L-segment phylogeny. The circular tree includes all LASV lineages (I-VI) represented by different color ranges. The squares at the tips represent host: empty square = human virus; black square = rodent virus; no square = laboratory virus. The outer circle represents geographic origin: dark grey = Nigeria, light grey: Mano River Union countries, intermediate grey = other (Togo, Benin & Mali). Newly sequenced viruses are depicted in red.

Figure 2: Lassa virus S-segment phylogeny and time-measured tree estimates. The circular tree includes all LASV lineages (I-VI) represented by different color ranges. The squares at the tips represent host: empty square = human virus; black square = rodent virus; no square = laboratory virus. The outer circle represents geographic origin: dark grey = Nigeria, light grey: Mano River Union countries, intermediate grey = other (Togo, Benin & Mali). Newly sequenced viruses are depicted in red. The time-measured tree includes all lineages except for lineage IV and V. Two clades are indicated that contain a pair of 2018 sequences. These clades and their timescale are shown in more detail on the right.
DISCLAIMER
The Irrua Specialist Teaching Hospital (ISTH), Edo State, Nigeria, Bernhard Nocht Institute for Tropical Medicine (BNITM), Hamburg, Germany, and Public Health England (PHE), UK, are committed to sharing data in public health emergencies. We release unpublished Lassa virus sequences from Nigeria to support the public health response as well as the development and evaluation of Lassa fever diagnostics or therapeutics. The data may be used and analyzed for these purposes. It is not permitted to use the sequences for publication, i.e. any type of communication with the general public that describes data generated with the use of the sequences. If you intend to so, please contact us directly.
Dr. Ephraim Epogbaini [email protected], Director of Institute of Lassa Fever Research and Control,
Prof. Stephan Günther [email protected], Director, WHO Collaborating Centre for Arboviruses and Hemorrhagic Fever Reference and Research, BNITM